Abu Simbel relief of Ramesses II shooting a composite bow at the Battle of Kadesh.
Computational chemistry, like all attempts to simulate reality, is defined by tradeoffs. Reality is far too complex to simulate perfectly, and so scientists have developed a plethora of approximations, each of which reduces both the cost (i.e. time) and the accuracy of the simulation. The responsibility of the practitioner is to choose an appropriate method for the task at hand, one which best balances speed and accuracy (or to admit that no suitable combination exists).
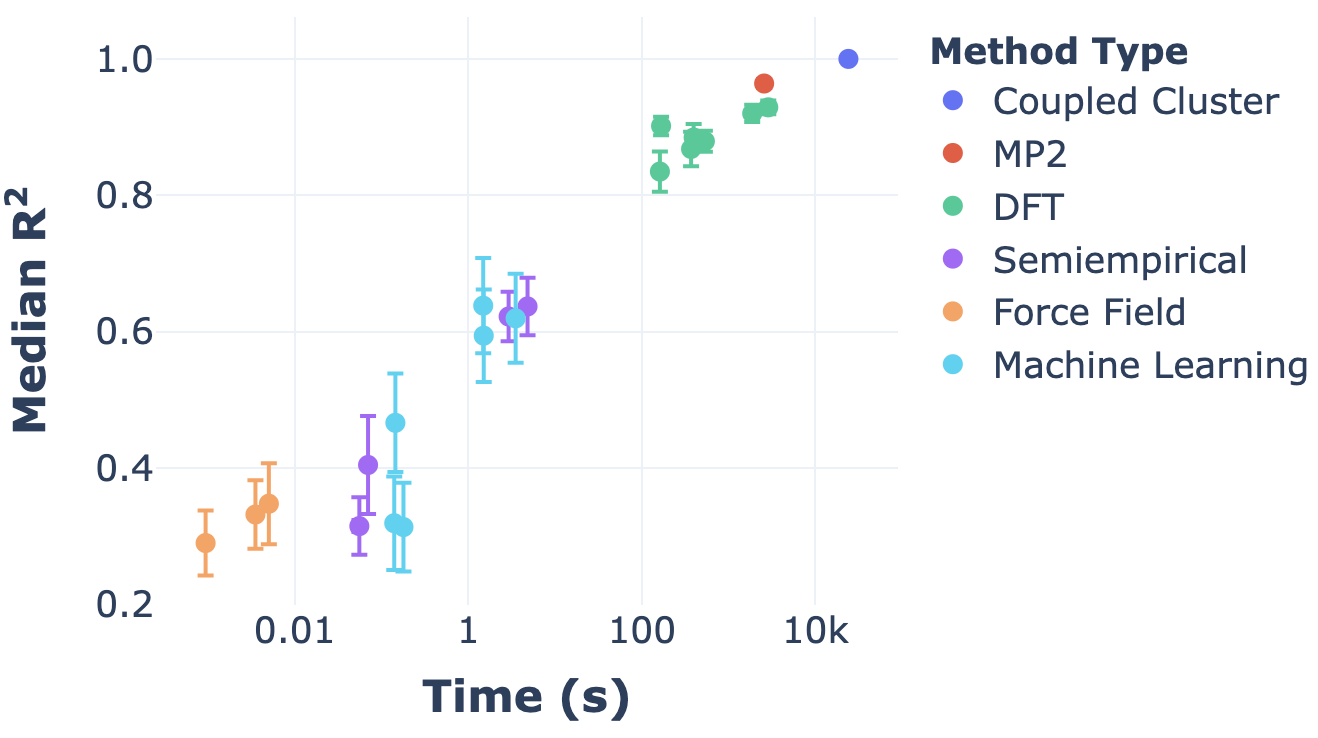
This situation can naturally be framed in terms of Pareto optimality: there’s some “frontier” of speed/accuracy combinations which are at the limit of what’s possible, and then there are suboptimal combinations which are inefficient. Here’s a nice plot illustrating exactly that, from Dakota Folmsbee and Geoff Hutchinson (ref):
The y axis represents R2 among different (relative) conformer energies, and the x axis is a log scale of computational time. The pattern shown here—exponential increases in time for linear increases in accuracy—is pretty common, unfortunately.
In this figure, the top left corner is the goal—perfect accuracy in no time at all—and the bottom right corner is the opposite. The diagonal line represents the Pareto frontier, and we can see that different levels of theory put you at different places along the frontier. Ab initio methods (DFT, MP2, coupled cluster) are slow but accurate, while force fields are fast but inaccurate, and semiempirical methods and ML methods are somewhere in the middle. (It’s interesting to observe that some ML methods are quite far from the optimal frontier, but I suppose that’s only to be expected from such a new field.)
An important takeaway from this graph is that some regions of the Pareto frontier are easier to access than others. Within e.g. DFT, it’s relatively facile to tune the accuracy of the method employed, but it’s much harder to find a method intermediate between DFT and semiempirical methods. (For a variety of reasons that I’ll write about later, this region of the frontier seems particularly interesting to me, so it’s not just an intellectual question.) This lacuna is what Stefan Grimme’s “composite” methods, the subject of today’s post, are trying to address.
I like to believe that these methods are named after the composite recurve bow, which is both smaller and more powerful than simple bows, but I don’t have evidence for this belief.
Pictured is Qing dynasty artwork of Zhang Xian shooting a composite bow.
What defines a composite method? The term hasn’t been precisely defined in the literature (as far as I’m aware), but the basic idea is to strip down existing ab initio electronic structure methods as much as possible, particularly the basis sets, and employ a few additional corrections to fix whatever inaccuracies this introduces. Thus, composite methods still have the essential form of DFT or Hartree–Fock, but rely heavily on cancellation of error to give them better accuracy than one might expect. (This is in contrast to semiempirical methods like xtb, which start with a more approximate level of theory and layer on a ton of corrections.)
Grimme and coworkers are quick to acknowledge that their ideas aren’t entirely original. To quote from their first composite paper (on HF-3c):
Several years ago Pople noted that HF/STO-3G optimized geometries for small molecules are excellent, better than HF is inherently capable of yielding. Similar observations were made by Kołos already in 1979, who obtained good interaction energies for a HF/minimal-basis method together with a counterpoise-correction as well as a correction to account for the London dispersion energy. It seems that part of this valuable knowledge has been forgotten during the recent “triumphal procession” of DFT in chemistry. The true consequences of these intriguing observations could not be explored fully at that time due to missing computational resources but are the main topic of this work.
And it’s not as though minimal basis sets have been forgotten: MIDIX still sees use (I used it during my PhD), and Todd Martinez has been exploring these ideas for a while. Nevertheless, composite methods seem to have attracted attention in a way that the above work hasn’t. I’ll discuss why this might be at the end of the post—but first, let’s discuss what the composite methods actually are.
HF-3c (2013)
HF-3c is a lightweight Hartree–Fock method, using a minimal basis set derived from Huzinaga’s MINIS basis set. To ameliorate the issues that Hartree–Fock and a tiny basis set cause, the authors layer in three corrections: the D3 dispersion correction (with Becke–Johnson damping), their recent “gCP” geometric counterpoise correction for basis set incompleteness error, and an “SRB” short-range basis correction for electronegative elements.
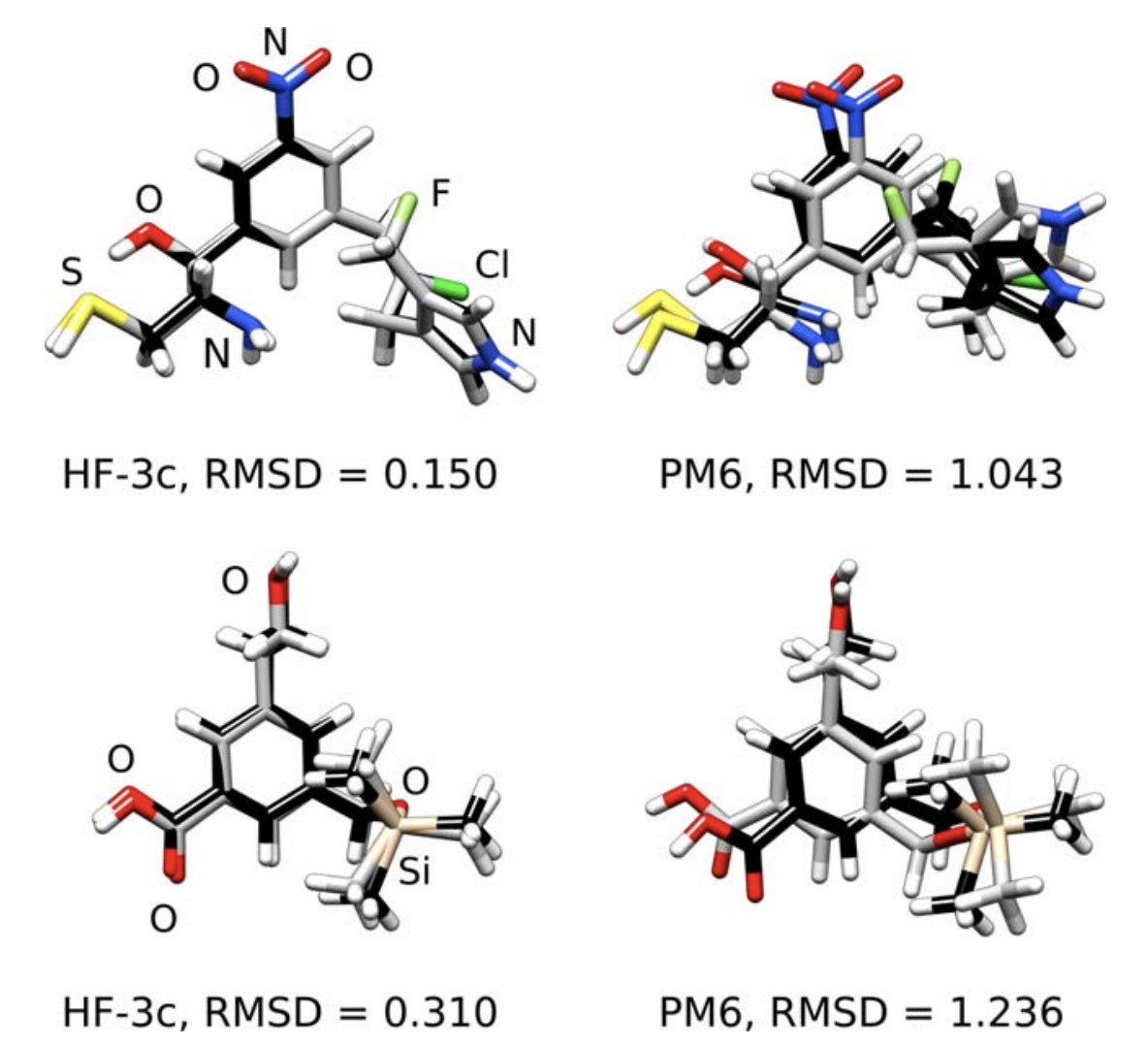
HF-3c is surprisingly good at geometry optimization and noncovalent interaction energies (MAE of 0.39 kcal/mol on the S66 benchmark set), works okay for dipole moments and vibrational frequencies, but seems bad for anything involving bond breaking. Thus the authors recommend it for optimization of ground-state complexes, and not so much for finding an entire potential energy surface.
Comparison of HF-3c and PM6 for geometry optimization, relative to B3LYP-D3/def2-TZVPP (black).
(One complaint about all of these papers: the basis set optimization isn’t described in much detail, and we basically have to take the authors’ word that what they came up with is actually the best.)
HF-3c(v) (2014)
HF-3c(v) is pretty much the same as HF-3c, but it uses a “large-core” effective core potential to describe all of the core electrons, making it valence-only. This makes it 2–4 times faster, but also seems to make it much worse than HF-3c: I’m not sure the speed is worth the loss in accuracy.
The authors only use it to explore noncovalent interactions; I’m not sure if others have used it since.
PBEh-3c (2015)
PBEh-3c is the next “3c” method, and the first composite DFT method. As opposed to the minimal basis set employed in HF-3c, Grimme et al here elect to use a polarized double-zeta basis set, which “significantly improves the energetic description without sacrificing the computational efficiency too much.” They settle on a variant of def2-SV(P) with an effective core potential and a few other modifications, which they call “def-mSVP.”
As before, they also add the D3 and gCP corrections (both slightly modified), but they leave out the SRB correction. The biggest change is that they also reparameterize the PBE functional, which introduces an additional four parameters: three in PBE, and one to tune the percentage of Fock exchange. The authors note that increasing the Fock exchange from 25% to 42% offsets the error introduced by basis set incompleteness.
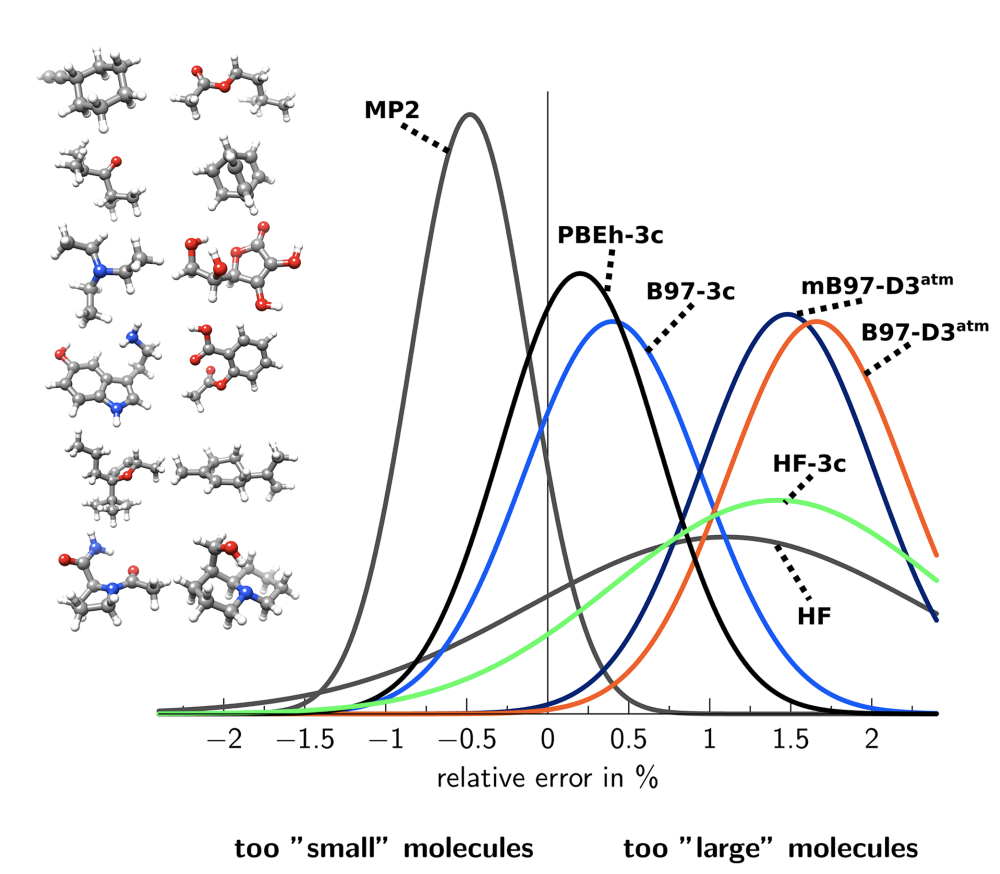
As before, the focus of the evaluation is on geometry optimization, and PBEh-3c seems to do very well (although not better than HF-3c on e.g. S66, which is surprising—the authors also don’t compare directly to HF-3c in the paper at all). PBEh-3c also does pretty well on the broad GMTKN30 database, which includes thermochemistry and barrier heights, faring just a bit worse than M06-2x/def2-SV(P).
HSE-3c (2016)
HSE-3c is basically the same as PBEh-3c, but now using a screened exchange variant to make it more robust and faster for large systems or systems with small band gaps. The authors recommend using PBEh-3c for small molecules or large band-gap systems, which is my focus here, so I won’t discuss HSE-3c further.
B97-3c (2018)
B97-3c is another DFT composite method, but it’s a bit different than PBEh-3c. PBE is a pretty simple functional with only three tunable parameters, while B97 is significantly more complex with ten tunable parameters. Crucially, B97 is a pure functional, meaning that no Fock exchange is involved, which comes with benefits and tradeoffs. The authors write:
The main aim here is to complete our hierarchy of “3c” methods by approaching the accuracy of large basis set DFT with a physically sound and numerically well-behaved approach.
For a basis set, the authors use a modified form of def2-TZVP called “mTZVP”, arguing that “as the basis set is increased to triple-ζ quality, we can profit from a more flexible exchange correlation functional.” This time, the D3 and SRB corrections are employed, but the gCP correction is omitted.
The authors do a bunch of benchmarking: in general, B97-3c seems to be substantially better than either PBEh-3c or HF-3c at every task, which isn’t surprising given the larger basis set. B97-3c is also often better than e.g. B3LYP-D3 with a quadruple-zeta basis set, meaning that it can probably be used as a drop-in replacement for most routine tasks.
Comparison of a few methods for geometry optimization, as assessed by rotational constants. Both B97-3c and PBEh-3c perform better than HF-3c.
B3LYP-3c (2020)
B3LYP-3c is a variant of PBEh-3c where you just remove the PBEh functional and replace it with B3LYP (without reparameterizing B3LYP at all). This is done to improve the accuracy for vibrational frequencies, since B3LYP performs quite well for frequencies. I’ve only seen this in one paper, so I’m not sure this will catch on (although it does seem to work).
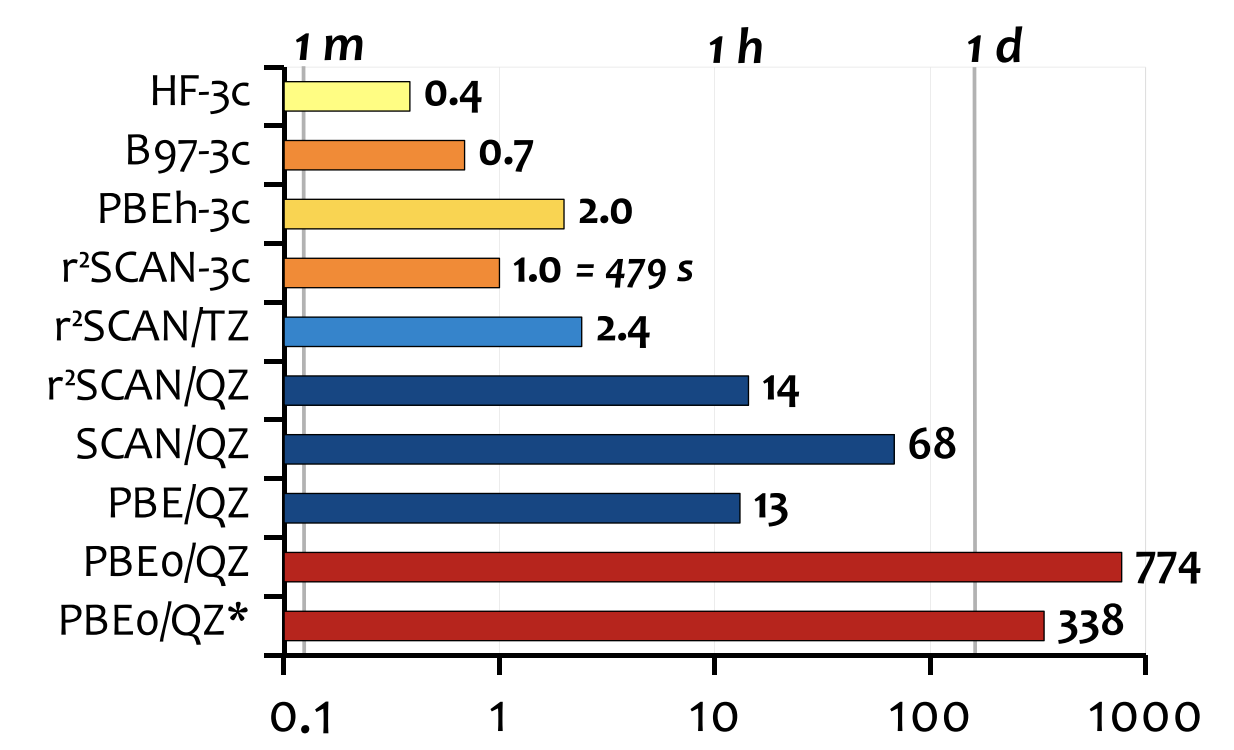
r2 SCAN-3c (2021)
Continuing our journey through the “Jacob’s ladder” of composite functionals, we arrive at r2 SCAN-3c, based on the meta-GGA r2 SCAN functional. No reparameterization of the base functional is performed, but the D4 and gCP corrections are added, and yet another basis set is developed: mTZVPP, a variant of the mTZVP basis set developed for B97-3c, which was already a variant of def2-TZVP.
The authors describe the performance of r2 SCAN-3c in rather breathless terms:
…we argue that r2 SCAN is the first mGGA functional that truly climbs up to the third rung of the Jacobs ladder without significant side effects (e.g., numerical instabilities or an overfitting behavior that leads to a bad performance for the mindless benchmark set).
…the new and thoroughly tested composite method r2 SCAN-3c provides benchmark-accuracy for key properties at a fraction of the cost of previously required hybrid/QZ approaches and is more robust than any other method of comparable cost. This drastically shifts the aforementioned balance between the computational efficiency and accuracy, enabling much larger and/or more thorough screenings and property calculations. In fact, the robustness and broad applicability of r2 SCAN-3c caused us to rethink the very structure of screening approaches.
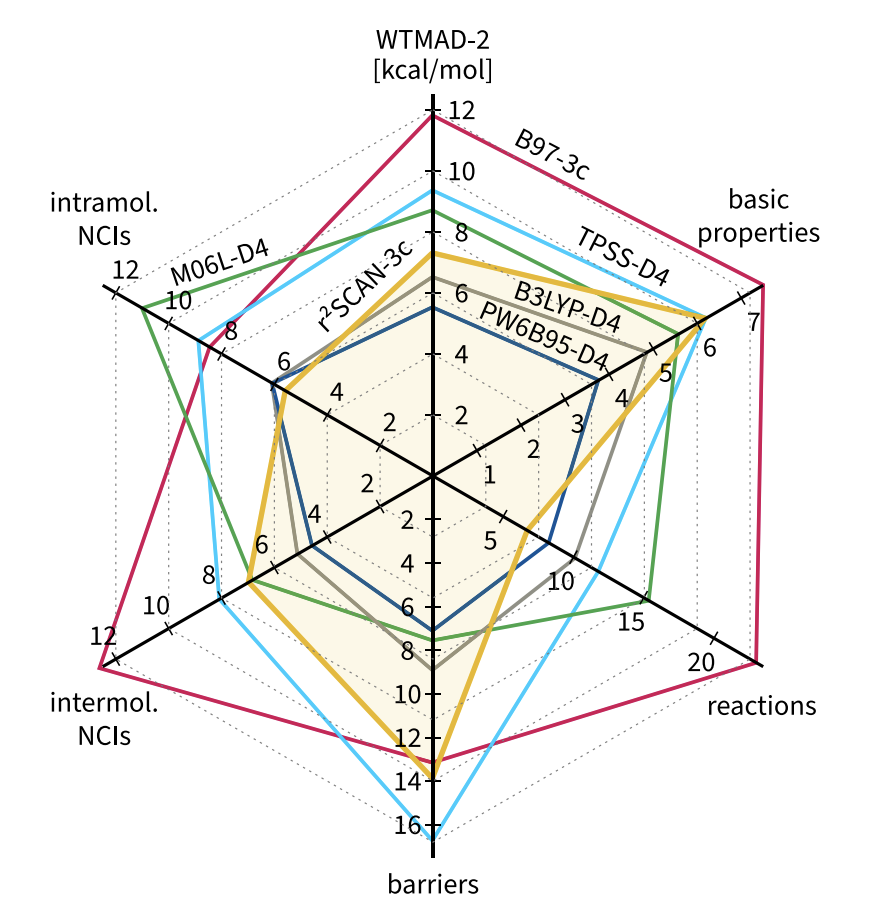
The amount of benchmarking performed is a little overwhelming. Here’s a nice figure that summarizes r2 SCAN-3c on the GMTKN55 database, and also compares it to B97-3c:
Notice how much better the results are than B97-3c.
And here’s a nice graph that shows time comparisons for a 153-atom system, which is something that’s obviously a key part of the value-add:
r2 SCAN-3c is only a bit slower than B97-3c, and both are substantially faster than PBEh-3c (probably because PBEh is a global hybrid). HF-3c, of course, is still fastest.
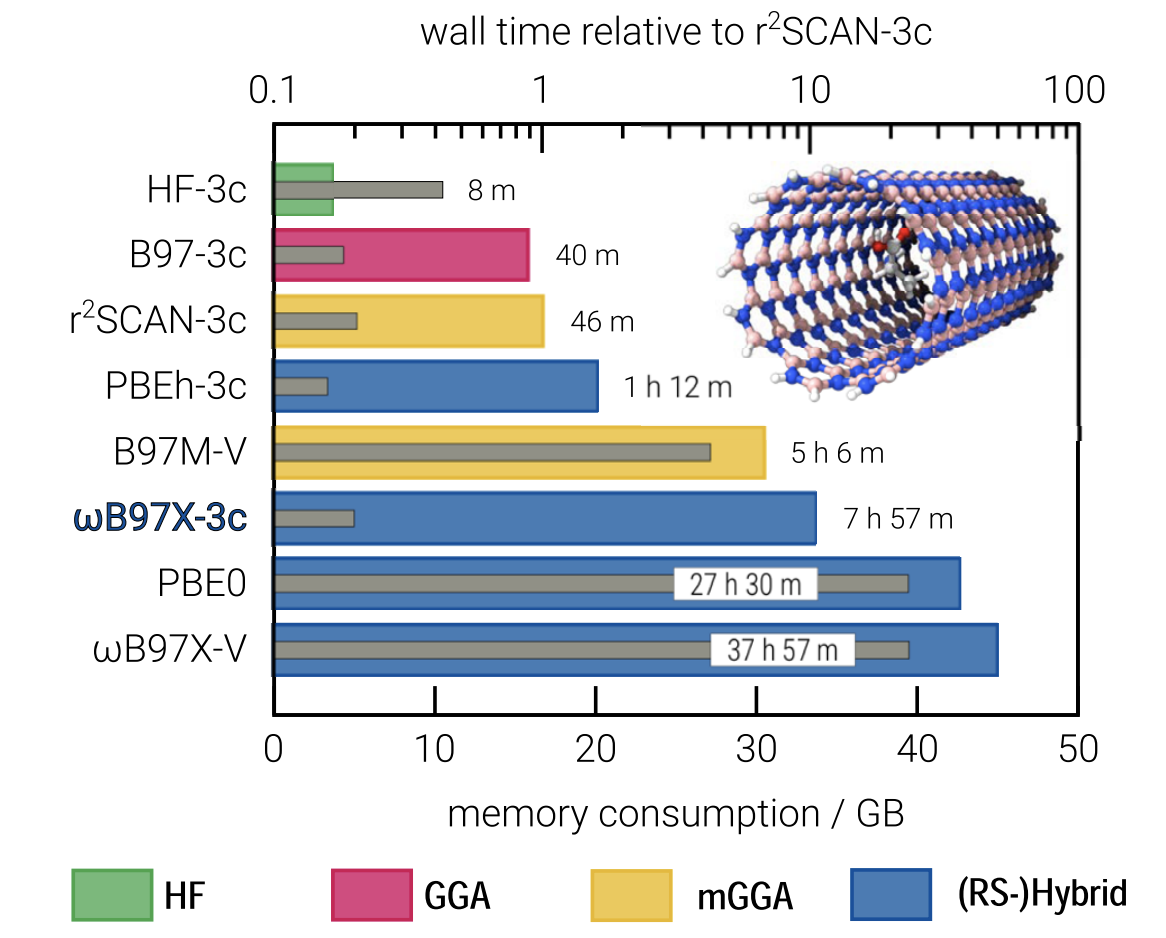
ωB97X-3c (2022)
Finally, we come to ωB97X-3c, a composite range-separated hybrid functional derived from Mardirossian and Head-Gordon’s ωB97X-V functional (which seems to me to be one of the best DFT methods, period). ωB97X-3c reintroduces Fock exchange, so it’s significantly more expensive than r2 SCAN-3c or B97-3c, but with this expense comes increased accuracy.
Interestingly, neither of the “weird” corrections (gCP or SRB) are employed for ωB97X-3c: it’s just an off-the-shelf unmodified functional, the now-standard D4 dispersion correction, and a specialized basis set. The authors acknowledge this:
Although ωB97X-3c is designed mostly in the spirit of the other “3c” methods, the meaning and definition of the applied “three corrections” have changed over the years. As before, the acronym stands for the dispersion correction and for the specially developed AO basis set, but here for the compilation of ECPs, which are essential for efficiency, as a third modification.
(But weren’t there ECPs before? Doesn’t even HF-3c have ECPs? Aren’t ECPs just part of the basis set? Just admit that this is a “2c” method, folks.)
The authors devote a lot of effort to basis-set optimization, because range-separated hybrids are so expensive that using a triple-zeta basis set like they did for B97-3c or r2 SCAN-3c would ruin the speed of the method. This time, they do go into more details, and emphasize that the basis set (“vDZP”) was optimized on molecules and not just on single atoms:
Molecule-optimized basis sets are rarely used in quantum chemistry. We are aware of the MOLOPT sets in the CP2K code and the polarization consistent (pc-n) basis sets by Jensen. In the latter, only the polarization functions are optimized with respect to molecular energies. A significant advantage of molecular basis set optimizations is that, contrary to atomic optimizations, all angular momentum functions (i.e., polarization functions not occupied in the atomic ground state) can be determined consistently, as already noted by VandeVondele and Hutter.
They also put together a new set of effective core potentials, which also helps to minimize the number of basis functions. Even so, ωB97X-3c is the slowest of the composite methods, as shown in this figure:
Slow, but still faster than the competition.
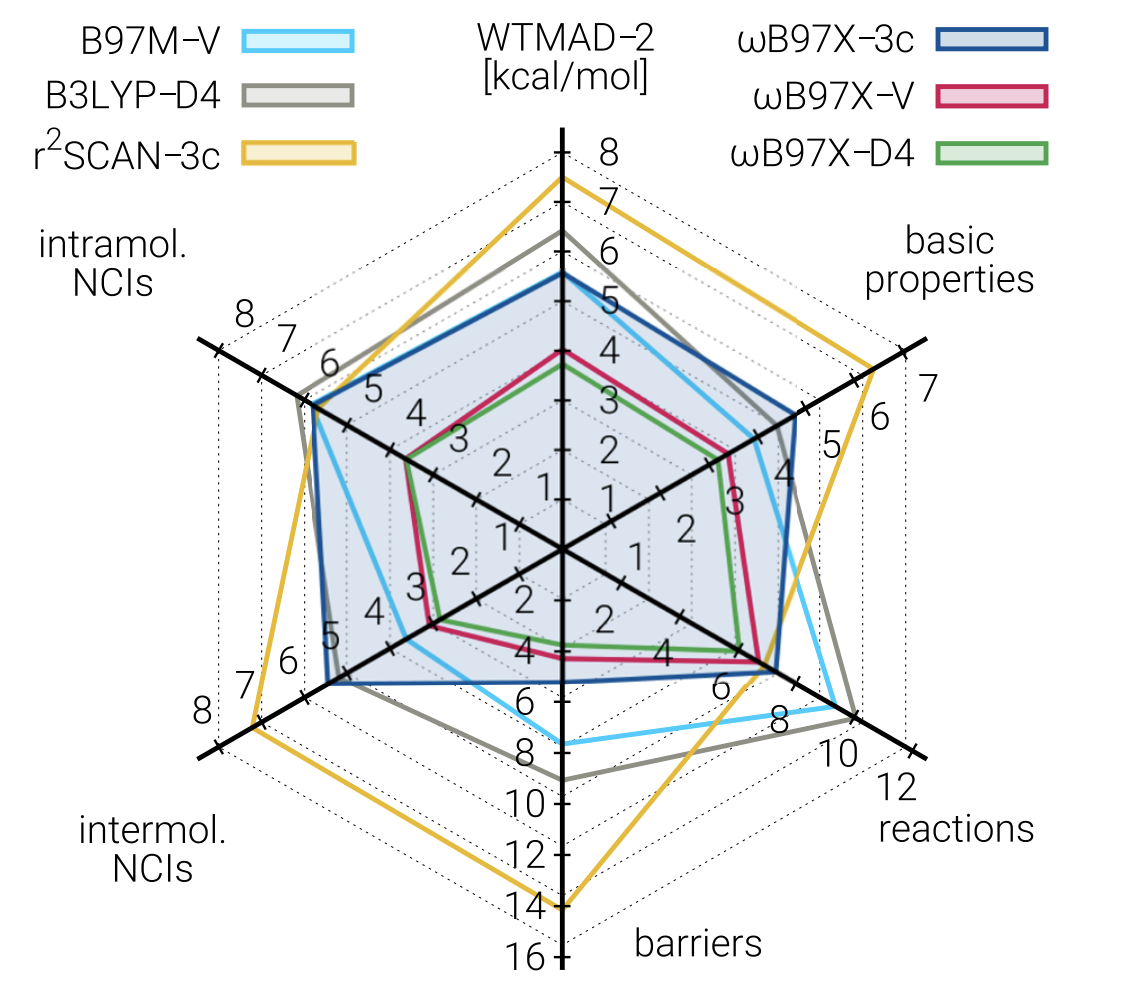
In terms of accuracy, ωB97X-3c is far better than r2 SCAN-3c, previously the best composite method, and is among the best-performing DFT methods in general for most benchmarks, although still outcompeted by ωB97X-V (and its close cousin ωB97X-D4, reparameterized in this work). The expense of ωB97X-3c means that for easy tasks like geometry optimization r2 SCAN-3c is still probably a better choice, but for almost anything else it seems that ωB97X-3c is an excellent choice.
An interesting observation is that ωB97X-3c is not just Pareto-optimal, but close to optimal in an absolute sense. I initially framed the goal of composite methods as finding new ways to increase speed while decreasing accuracy: but here it seems that we can gain a ton of speed without losing much accuracy at all! This should be somewhat surprising, and the implications will be discussed later.
Overall Summary
Excluding some of the more specific composite methods, there seem to be three tiers here:
HF-3c, which is very fast but only reliable for geometry optimizations and pretty easy property calculations (e.g. dipole moment, rough frequencies). But also, geometry optimizations are ubiquitous, so this is still very important.
PBEh-3c, B97-3c, and r2 SCAN-3c, which are all roughly the same speed despite substantial differences. Of these, PBEh-3c seems like the slowest and least accurate, and I’d be tempted to use either of the others first, especially r2 SCAN-3c. (I’m curious about the tradeoffs that a mGGA functional has versus a GGA functional like B97-3c—are there pathological integration grid behaviors that B97-3c avoids? See Figure 6 here.)
ωB97X-3c, which seems to be in a class of its own, and better than almost everything else (not just other composites). That being said, it’s still new, and no one loves a method better than its own authors. We’ll have to see some real-world tests to make sure things are as promising as they seem.
Conclusions
After decades of theorists mocking people for using small basis sets, it’s ironic that intentionally embracing cancellation of error is trendy again. I’m glad to see actual theorists turn their attention to this problem: people have never stopped using “inadequate” basis sets like 6-31G(d), simply because nothing larger is practical for many systems of interest!
The results from this body of work suggest that current basis sets are far from optimal, too. The only piece of ωB97X-3c that’s new is the basis set, and yet that seems to make a huge difference relative to state-of-the-art. What happens if vDZP is used for other methods, like B97? The authors suggest that it might be generally effective, but more work is needed to study this further.
Update: Jonathon Vandezande and I investigated this question, and it turns out vDZP is effective with other density functionals too. You can read our preprint here!
Basis-set optimization seems like a “schlep” that people have avoided because of how annoying it is, or something which is practically useful but not very scientifically interesting or publishable. Perhaps the success of composite methods will push more people towards studying basis sets; I think that would be a good outcome of this research. It seems unlikely to me that vDZP cannot be optimized further; if the results above are any indication, the Pareto frontier of basis sets can be advanced much more.
I’m also curious if Grimme and friends would change HF-3c and the other early methods, knowing what they know now. Do better basis sets alleviate the need for gCP and SRB, or is that not possible with a minimal basis set? What about D3 versus D4 (which wasn’t available at the time)? Hopefully someone finds the time to go back and do this, because to my knowledge HF-3c sees a good amount of use.
Perhaps my favorite part of this work, though, is the ways in which composite methods reduce the degrees of freedom available to end users. “Classic” DFT has a ton of tunable parameters (functional, basis set, corrections, solvent models, thresholds, and so forth), and people frequently make inefficient or nonsensical choices when faced with this complexity. In contrast, composite methods make principled and opinionated choices for many of these variables, thus giving scientists a well-defined menu of options.
This also makes it easier for outsiders to understand what’s going on. I wrote about this over a year ago:
While a seasoned expert can quickly assess the relative merits of BYLP/MIDI! and DSD-PBEP86/def2-TZVP, to the layperson it’s tough to guess which might be superior… The manifold diversity of parameters employed today is a sign of [computational chemistry]’s immaturity—in truly mature fields, there’s an accepted right way to do things.
The simplicity of composite methods cuts down on the amount of things that you have to memorize in order to understand computational chemistry. You only have to remember “HF-3c is fast and sloppy,” rather than trying to recall how many basis functions pcseg-1 has or which Minnesota functional is good for main-group geometries.
So, I’m really optimistic about this whole area of research, and I’m excited that other labs are now working on similar things (I didn’t have space to cover everyone’s contributions, but here’s counterpoise work from Head-Gordon and atom-centered potentials from DiLabio). The next challenge will be to get these methods into the hands of actual practitioners…
ICYMI: Ari and I announced our new company, Rowan! We wrote an article about what we're hoping to build, which you can read here. Also, this blog is now listed on The Rogue Scholar, meaning that posts have DOIs and can be easily cited.
Conventional quantum chemical computations operate on a collection of atoms and create a single wavefunction for the entire system, with an associated energy and possibly other properties. This is great, but sometimes we want to understand things in more detail. For instance, if we have a host A and two guests Bgood and Bbad, a normal calculation would just tell us that E(A•Bgood) is lower than E(A•Bbad), without giving any clue as to why.
Enter EDA. EDA, or “energy decomposition analysis,” is a family of techniques used to dissect interactions in a system with multiple molecules. In this case, an EDA calculation on the AB system would break down the interaction between A and B into various components, which could be used to help scientists understand the origin of the difference, and perhaps used for continued molecular design.
Unfortunately, EDA has always seemed like a pretty troubled technique to me. Wavefunctions are inherently not localized to individual fragments of a multimolecular system—you can’t just slice apart the molecular orbitals or the density matrix and end up with anything that’s physically sane. So you have to do some computational gymnastics to get energetic terms which are at all meaningful. Many such gymnastic workflows have been proposed, leading to a veritable alphabet soup of different EDA methods.
(It’s worth skimming this review on different EDA methods to get a sense for some of the questions the field faces, and also to laugh at how Alston Misquitta & Krzysztof Szalewicz use the review as a chance to relentlessly advertise SAPT and denigrate any and all competing methods.)
I’ll briefly outline how the EDA-NCOV method works for a system AB (following this review), to give a sense for the flavor of the field:
Optimized ground-state fragments A0 and B0 are distorted to the geometries and electronic states (A & B) which they possess in AB, and the energy required for this distortion/excitation is termed Eprep. (The difference between E(AB) and E(A) + E(B) is called Eint, and the total binding energy is equal to Eint + Eprep.)
The distorted fragments A and B are brought together (with frozen charge densities) to form the “promolecule” AB0, and the change in energy is termed Eelstat, the quasiclassical Coulomb interaction energy (typically attractive). The wavefunction for AB0 is ΨAΨB.
The product wavefunction ΨAΨB is antisymmetrized and renormalized to give an “intermediate state” Ψ0 with energy E0, and the change in energy is termed EPauli, originating from Pauli repulsion. This component is always repulsive.
Ψ0 is relaxed to yield the final wavefunction ΨAB. The change in energy is termed Eorb, because it arises from orbital interactions, and is always attractive.
Thus, Eint = Eelstat + EPauli + Eorb. (Dispersion can also be added if an exogenous dispersion correction is employed—that’s pretty trivial.)
The critical reader might observe that the steps taken to obtain these numbers are pretty odd, and that the components of the interaction energy arise from differences in energy between bizarre nonphysical states. Thus, the interpretation of terms like Eelstat in terms of actual physical interactions might not be as easy as it seems. The authors of the above review agree:
It is important to realize that the identification of the three major terms ΔEelstat, ΔEPauli, and ΔEorb with specific interactions is conceptually attractive but must not be taken as genuine expression of the physical forces.
Unfortunately, it seems that imprecise concepts familiar to experimental chemists like “steric repulsion” and “electrostatic attraction” have to be discarded in favor of precise terms like EPauli. Too bad they’re virtually uninterpretable!
And what’s worse is that different EDA-type schemes don’t even give the same results. A paper out today in JACS from Zare/Shaik discusses the use of EDA and related schemes in studying the origin of the hydrogen bond (a pretty fundamental question), motivated by the substantial disagreement between various techniques:
It is important to realize that different methods (e.g., BOVB, ALMO-EDA, NEDA, and BLW) do not fully agree with one another about whether the dominant stabilizing term is ΔEPOL or ΔECT in a particular HB.
While the authors make a good case that the sum of these two terms is relatively conserved across methods, and that it’s this term that we should care about for hydrogen bonds, the conclusions for EDA broadly are not encouraging. (Note, too, that EPOL and ECT don’t even appear in the EDA-NCOV method summarized above—another reason that EDA is a frustrating field!)
And even if the theorists eventually put their heads together and develop a version of EDA that doesn’t have these pitfalls, it’s still not clear that any form of EDA will give the answers that experimental chemists are looking for. Chemistry is complicated, and ground- or transition-state structures arise from a delicate equilibrium between opposing factors: steric repulsion, electrostatic attraction, bond distances, torsional strain, dispersion, &c.
As a result, one can see large changes in the contribution of individual factors even while the overall structure’s stability is minimally perturbed (enthalpy–entropy compensation is a classic example, as is Fig. 2 in this review on distortion–interaction analysis). Looking only at changes in individual factors isn’t always a useful way to gain insight from computation.
For example, imagine a nucleophile adding to two faces of an oxocarbenium, a bulky face and an unhindered face. Based on this description, we might expect to see that TSbulky has higher steric repulsion than TSunhindered (if we’re lucky enough to find a way to extract Esteric out of our EDA method). But it’s also likely that the nucleophile might take a less favorable trajectory towards the oxocarbenium in TSbulky to avoid steric repulsion, thus weakening key orbital interactions. These changes might even end up being larger in magnitude than the destabilization induced by steric repulsion. Is the correct answer, then, that TSbulky is higher in energy because of decreased Eorb, not increased Esteric?
The solution is to recognize that causation is not unique (cf. Aristotle), and so there’s no one right answer here. Within the constraints of the EDA framework, the theorist wouldn’t be incorrect in saying that Eorb is the driving factor—but the experimental chemist might reasonably expect “the bulky TS is destabilized by steric repulsion” as their answer, since this is the root cause of the changes between the two structures. (I side with the experimentalists here.)
And the precisely defined concepts favored by theorists are often hard for experimental scientists to work with. Even if the correct answer in the above scenario were “TSbulky is destabilized by decreased orbital overlap”—what’s an experimentalist supposed to do with this information, add more orbitals? (This is how I feel about Trevor Hamlin’s work on Pauli repulsion.) The steric explanation at least suggests an intuitive solution: make the bulky group or the nucleophile smaller. If the purpose of EDA is to help people to understand intermolecular interactions better on a conceptual level, I’m not sure it’s succeeding in most cases.
(The only use of EDA that led to an actual experimental advance which I’m aware of is Buchwald/Peng Liu’s body of work on ligand–substrate dispersion in hydrocupration: study, new ligand, ligand from Hartwig. I don’t think it’s a coincidence that these papers focus on dispersion, one of the easiest pieces of EDA to decouple and understand.)
I don’t mean to be too critical here. The ability to break intermolecular interactions down into different components is certainly useful, and it seems likely that some version of EDA will eventually achieve consensus and emerge as a useful tool. But I think the utility of EDA even in the best case is pretty limited. Quantum chemistry is complicated, and if we think we can break it down into easy-to-digest components and eliminate the full nonlocal majesty of the Schrodinger equation, we’re lying to ourselves (or our experimental collaborators). Compute with caution!
Recently in off-blog content: I coauthored an article with Eric Gilliam on how LLMs can assist in hypothesis generation and help us all become more like Sharpless. Check it out!
The Pauling model for enzymatic catalysis states that enzymes are “antibodies for the transition state”—in other words, they preferentially bind to the transition state of a given reaction, rather than the reactants or products. This binding interaction stabilizes the TS, thus lowering its energy and accelerating the reaction.
This is a pretty intuitive model, and one which is often employed when thinking about organocatalysis, particularly noncovalent organocatalysis. (It’s a bit harder to use this model when the mechanism changes in a fundamental way, as with many organometallic reactions.) Many transition states have distinctive features, and it’s fun to think about what interactions could be engineered to recognize and stabilize only these features and nothing else in the reaction mixture.
(For instance, chymotrypsin contains a dual hydrogen-bond motif called the “oxyanion hole” which stabilizes developing negative charge in the Burgi–Dunitz TS for alcoholysis of amides. The negative charge is unique to the tetrahedral intermediate and the high-energy TSs to either side, so reactant/product inhibition isn’t a big issue. This motif can be mimicked by dual hydrogen-bond donor organocatalysts like the one my PhD lab specialized in.)
The downside of this approach to catalyst design is that each new sort of reaction mechanism requires a different sort of catalyst. One TS features increasing negative charge at one place, while another features increasing positive charge at another, and a third is practically charge-neutral the whole way through. What if there were some feature that was common to all transition states?
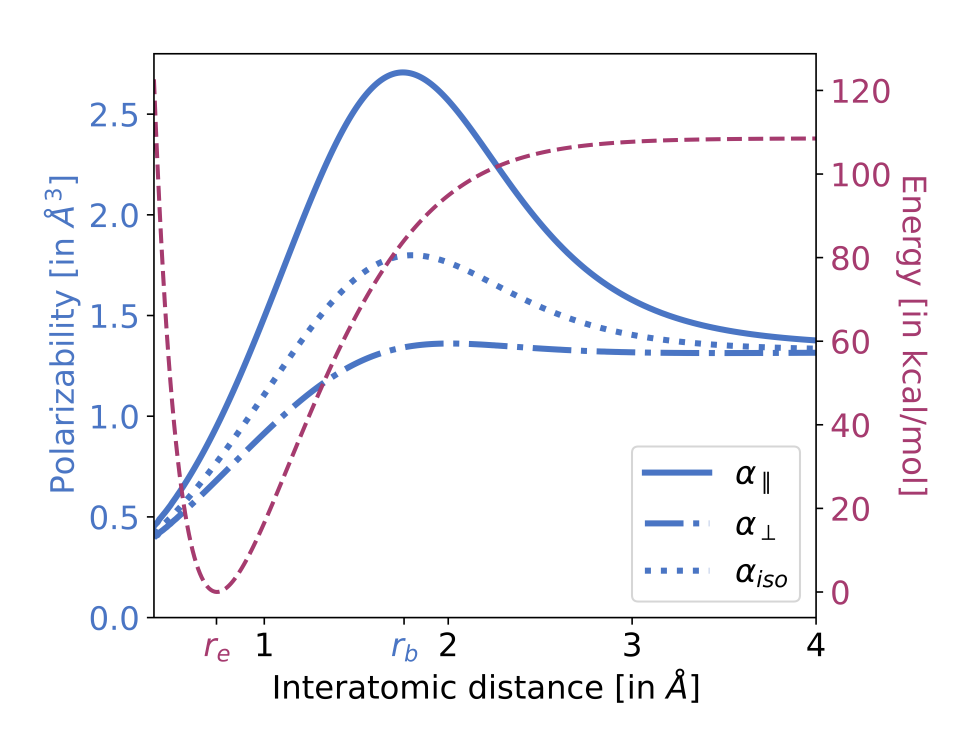
A recent preprint from Diptarka Hait and Martin Head-Gordon suggests an interesting answer to this question: polarizability. (Diptarka, despite just finishing his PhD, is a prolific scientist with a ton of different interests, and definitely someone to keep an eye on.) The authors tackle the question of when precisely a stretching bond can be considered “broken.” An intuitive answer might be “the transition state,” but as the paper points out, plenty of bond-breaking potential energy surfaces lack clear transition states (e.g. H2, pictured below in red).
Hydrogen PES (red), with various polarizability components shown in blue.
Instead, the authors propose that polarizability is a good way to study this question. As seen in the following graph, polarizability (particularly α||, the component parallel to the bond axis) first increases as a bond is stretched, and then decreases, with a sharp and easily identifiable maximum about where a bond might be considered to be broken. This metric tracks with the conventional PES metric in cases where the PES is well-defined (see Fig. 5), which is comforting. Why does this occur?
The evolution of α [polarizability] can be rationalized in the following manner. Upon initially stretching H2 from equilibrium, the bonding electrons fill up the additional accessible volume, resulting in a more diffuse (and thus more polarizable) electron density. Post bond cleavage however, the electrons start localizing on individual atoms, leading to a decrease in polarizability.
In other words, electrons that are “caught in the act” of reorganizing between different atoms are more polarizable, whereas electrons which have settled into their new atomic residences are less polarizable again.
This is cool, but how can we apply this to catalysis? As it happens, there are already a few publications (1, 2) from Dennis Dougherty and coworkers dealing with exactly this question. They show that cyclophanes are potent catalysts for SN2-type reactions that both create and destroy cations, and argue that polarizability, rather than any charge-recognition effect, undergirds the observed catalysis:
Since transition states have long, weak bonds, they are expected to be more polarizable than ground-state substrates or products. The role of the host is to surround the transition state with an electron-rich-system that is polarizable and in a relatively fixed orientation, so that induced dipoles in both the host and the transition state are suitably aligned… Note that in this model, it is not sufficient that polarizability contributes to binding. Polarizability must be more important for binding transition states than for binding ground states. Only in this way can it enhance catalysis.
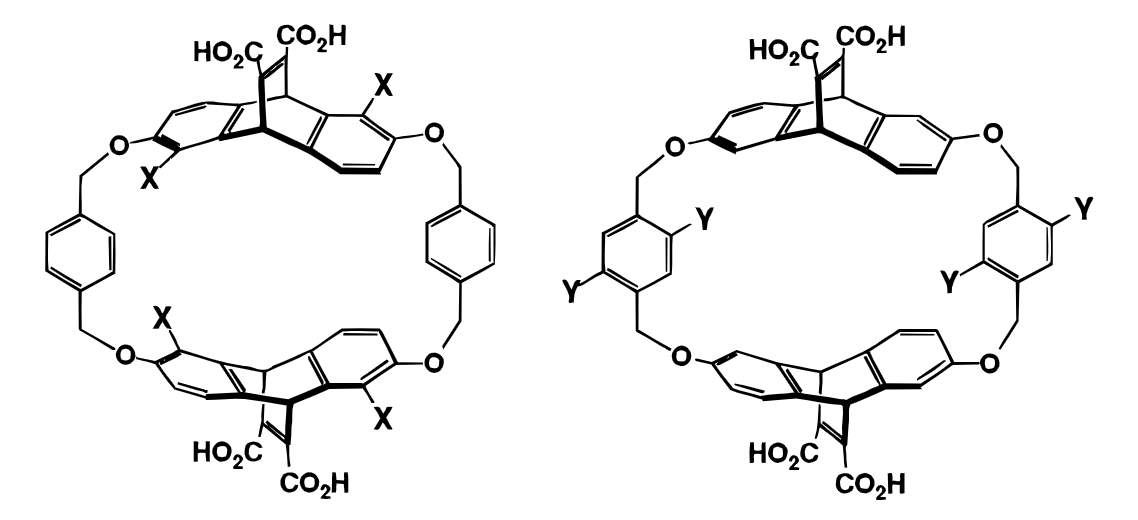
To defend this argument, the authors prepare a series of variously substituted cyclophanes, and show that while Hammett-type electronic tuning of the aryl rings has relatively small effects, adding more heavy atoms always increases the rate, with the biggest effect observed with bromine (the heaviest element attempted). Heavier atoms are more polarizable, so this supports the argument that polarizability, rather than any specific electrostatic effect, is responsible for catalysis.
The cyclophanes used as catalysts: Y=Br is the best.
The Dougherty work is performed on a very specific model system, and the absolute rate accelerations seen aren’t massive (about twofold increase relative to the protio analog), so it’s not clear that this will actually be a promising avenue for developing mechanism-agnostic catalysts.
But I think this line of research is really interesting, and merits further investigation. Pericyclic reactions, which involve large electron clouds and multiple forming–breaking bonds and often feature minimal development of partial charges, seem promising targets for this sort of catalysis—what about the carbonyl–ene reaction or something similar? The promise of new catalytic strategies that complement existing mechanism-specific interactions is just too powerful to leave unstudied.
Thanks to Joe Gair for originally showing me the Dougherty papers referenced.
Since the ostensible purpose of organic methodology is to develop reactions that are useful in the real world, the utility of a method is in large part dictated by the accessibility of the starting materials. If a compound is difficult to synthesize or hazardous to work with, then it’s difficult to convince people to use it in a reaction (e.g. most diazoalkanes). Organic chemists are pragmatic, and would usually prefer to run a reaction that starts from a commercial and bench-stable starting material.
For instance, this explains the immense popularity of the Suzuki reaction: although the Neigishi reaction (using organozinc nucleophiles) usually works better for the same substrates, you can buy lots of the organoboron nucleophiles needed to run a Suzuki and leave them lying around without taking any precautions. In contrast, organozinc compounds usually have to be made from the corresponding organolithium/Grignard reagent and used freshly, which is considerably more annoying.
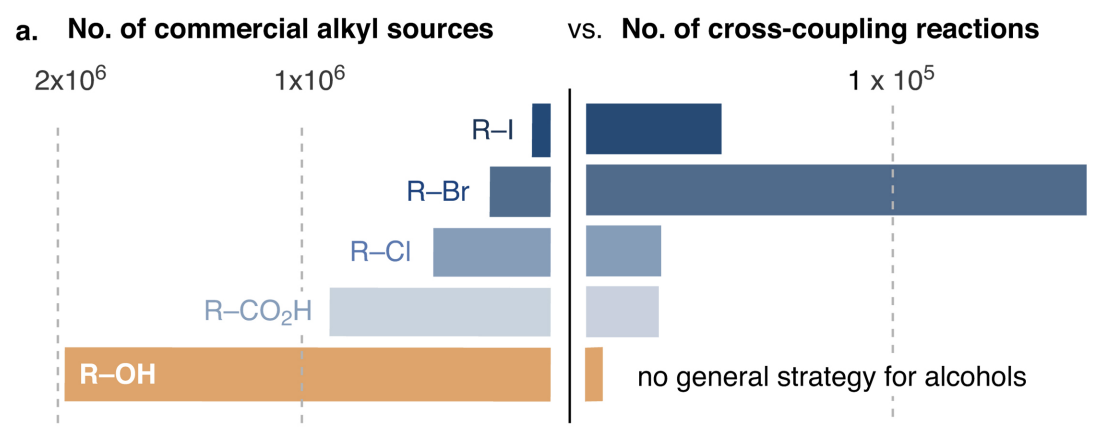
The ideal starting material, then, is one which is commercially available and cheap. In recent years, it’s become popular to advertise new synthetic methods by showing that they work on exceptionally cheap and common functional groups, and in particular to compare the abundance of different functional groups to demonstrate that one starting material is more common than another. To pick just one of many examples, Dave MacMillan used this plot to show why cross-coupling reactions of alcohols were important (ref):
This visual works really well.
When I saw MacMillan’s talk at MIT last year, I was curious what it would take to make additional graphics like this. The “number of reactions” plot can be made pretty easily from Reaxys, but I’ve always been uncertain how the “number of commercial sources” plots are made: I haven’t seen references listed for these numbers, nor is anything usually found in the Supporting Information.
I decided to take a swing at getting this data myself by analyzing the Mcule "building blocks" database, which contains about 3.5 million compounds. Although Mcule doesn't define what a building block is (at least, not that I can find), it’s likely that their definition is similar to that of ZINC, which defines building blocks as “those catalogs of compounds available in preparative quantities, typically 250 mg or more” (ref). This seems like a reasonable proxy for the sorts of compounds synthetic chemists might use in reactions. I defined patterns to match a bunch of functional groups using SMARTS/SMILES, and then used RDKit to find matches in the Mcule building blocks database. The code can be found on Github, along with the patterns I used.
The results are shown below. As expected, ethers, amines, amides, and alcohols are quite common. Surprisingly, aryl chlorides aren't that much more common than aryl bromides—and, except for aliphatic fluorides, all aliphatic halides are quite rare. Allenes, carbodiimides, and SF5 groups are virtually unheard of (<100 examples).
Functional Group
Number
Percent
acid chloride
6913
0.19
alcohol
1022229
28.60
aliphatic bromide
42018
1.18
aliphatic chloride
70410
1.97
aliphatic fluoride
650576
18.20
aliphatic iodide
3159
0.09
alkene
176484
4.94
alkyne
35577
1.00
allene
99
0.00
amide
518151
14.50
anhydride
1279
0.04
aryl bromide
451451
12.63
aryl chloride
661591
18.51
aryl fluoride
618620
17.31
aryl iodide
216723
6.06
azide
5164
0.14
aziridine
748
0.02
carbamate
127103
3.56
carbodiimide
28
0.00
carbonate
1231
0.03
carboxylic acid
410860
11.49
chloroformate
250
0.01
cyclobutane
195728
5.48
cyclopropane
349455
9.78
diene
10188
0.29
difluoromethyl
163395
4.57
epoxide
5859
0.16
ester
422715
11.83
ether
1434485
40.13
isocyanate
1440
0.04
isothiocyanate
1389
0.04
nitrile
209183
5.85
nitro
126200
3.53
pentafluorosulfanyl
18
0.00
primary amine
904118
25.29
secondary amine
857290
23.98
tertiary amine
609261
17.04
trifluoromethoxy
18567
0.52
trifluoromethyl
455348
12.74
urea
518151
14.50
Total
3574611
100.00
(Fair warning: I’ve spotchecked a number of the SMILES files generated (also on Github), but I haven’t looked through every molecule, so it’s possible that there are some faulty matches. I wouldn’t consider these publication-quality numbers yet.)
An obvious caveat: there are lots of commercially “rare” functional groups which are easily accessible from more abundant functional groups. For instance, acid chlorides seem uncommon in the above table, but can usually be made from ubiquitous carboxylic acids with e.g. SOCl2. So these data shouldn’t be taken as a proxy for a more holistic measure of synthetic accessibility—they measure commercial availability, that’s all.
What conclusions can we draw from this?
The most common functional groups are the milquetoast ones: alcohols, amines, esters, etc. Perhaps this explains where all the new reactions have gone: unless your new method works on alcohols or amines, it will struggle to get traction in most of chemical space relative to e.g. Williamson ether synthesis or reductive amination. (Kudos to MacMillan for identifying this; vide supra.)
Ureas are much more common than you’d expect from academic methods papers. This I think speaks to the difference between what methodologists want and what medicinal chemists want. Ureas are a bit annoying to work with: they’re pretty polar by the standards of academia, they’re not always soluble in organic solvents, and they have a tendency to stick to transition metal catalysts or get deprotonated by strong bases. But they’re easy to make in libraries, since the isocyanate/amine disconnection is so robust, and they’re excellent hydrogen-bond donors and acceptors.
CORRECTION: There's a SMARTS error, so the match for "ureas" actually matches amides—disregard this section. Thanks to @wmdhn for catching this.
Uncommon functional groups, like SF5 and allenes, are very uncommon. If you want to introduce an SF5 group, you are in for a rough time: there aren’t great ways to add it to molecules (although there have been some steps forward in recent years), and there are only 18 commercial examples. So people can write as many papers as they want about how cool SF5 groups are: I still doubt we’ll see them used very much in the near future.
But also, the abundance of a given functional group is very elastic in the long run. Trifluoromethyl groups used to be extremely rare—they’re not found in nature!—but now 1 in 8 molecules has a CF3 group. CF3 just turns out to be a very good handle for a lot of molecular design tasks, and so people found ways to introduce it all over the place, and now it’s not hard to get molecules that have trifluoromethyl groups. Synthetic chemists should feel good about this.
The functional-group-specific SMILES files are in the previously mentioned Github repo, so anyone who wants to e.g. look through all the commercially available alkenes and perform further cheminformatics analyses can do so. I hope the attached code and data helps other chemists perform similar, and better, studies, and that this sort of thinking can be useful for those who are currently engaged in reaction discovery.
Thanks to Eric Jacobsen for helpful conversations about these data.
In his fantastic essay “The Two Cultures,” C. P. Snow observed that there was (in 1950s England) a growing divide between the academic cultures of science and the humanities:
Literary intellectuals at one pole—at the other scientists, and as the most representative, the physical scientists. Between the two a gulf of mutual incomprehension—sometimes (particularly among the young) hostility and dislike, but most of all lack of understanding. They have a curious distorted image of each other. Their attitudes are so different that, even on the level of emotion, they can't find much common ground.
He reflects on the origins of this phenomenon, which he contends is new to the 20th century, and argues that it ought to be opposed:
This polarisation is sheer loss to us all. To us as people, and to our society. It is at the same time practical and intellectual and creative loss, and I repeat that it is false to imagine that those three considerations are clearly separable. But for a moment I want to concentrate on the intellectual loss.
Snow’s essay is wonderful: his portrait of a vanishing cultural intellectual unity should inspire us all, scientists and otherwise, to improve ourselves, and the elegiac prose reminds the reader that even the best cultural institutions are fragile and fleeting things.
I want to make an analogous—but much less powerful—observation about the two cultures present in atomistic simulation. I’ll call these the “QM tribe” and the “MD tribe” for convenience: crudely, “people who use Gaussian/ORCA/Psi4 for their research” and “people who use Schrodinger/AMBER/OpenMM/LAMMPS for their research,” respectively. Although this dichotomy is crude, I contend there are real differences between these two groups, and that their disunity hurts scientific progress.
The Nature of Energy Surfaces
The most fundamental disagreement between these two cultures is in how they think about energy surfaces, I think. Most QM-tribe people think in terms of optimizing to discrete critical points on the potential energy surface: one can perform some sort of gradient-informed optimization to a ground state, or follow negative eigenvalues to a transition state.
Implicit to this assumption is that there exist well-defined critical points on the PES, and that finding such critical points is meaningful and productive. Conformers exist, and many people now compute properties as Boltzmann-weighted averages over conformational ensembles, but this is usually done for 10–100 conformers, not thousands or millions. Entropy and solvation, if they’re considered at all, are viewed as corrections, not key factors: since QM is so frequently used to study high-barrier bond-breaking processes where enthalpic factors dominate, one can often get reasonable results with cartoonish treatments of entropy.
In contrast, MD-tribe scientists generally don’t think about transition states as specific configurations of atoms—rather, a transition state can emerge from some sort of simulation involving biased sampling, but it’s just a position along some abstract reaction coordinate, rather than a structure which can be visualized in CYLView. Any information gleaned is statistical, rather than concretely visual (e.g. “what is the mean number of hydrogen bonds to this oxygen near this transition state”).
Unlike the QM tribe, MD-tribe scientists generally cannot study bond-breaking processes, and so focus on conformational processes (protein folding, aggregation, nucleation, transport) where entropy and solvation are of critical importance: as such, free energy is almost always taken into consideration by MD-tribe scientists, and the underlying PES itself is rarely (to my knowledge) viewed as a worthy topic of study in and of itself.
Molecular Representations
This divide also affects how the two cultures view the task of molecular representation. QM-tribe scientists generally view a list of coordinates and atomic numbers as the most logical representation of a molecule (perhaps with charge and multiplicity information). To the QM tribe, a minimum on the PES represents a structure, and different minima naturally ought to have different representations. Bonding and bond order are not specified, because QM methods can figure that out without assistance (and it’s not uncommon for bonds to change in a QM simulation anyway).
In contrast, people in the MD tribe generally want a molecular representation that’s independent of conformation, since many different conformations will intrinsically be considered. (See Connor Coley’s presentation from a recent MolSSI workshop for a discussion of this.) Thus, it’s common to represent molecules through their topology, where connectivity and bond order are explicitly specified. This allows for some pretty wild simulations of species that would be reactive in a QM simulation, but also means that e.g. tautomers can be a massive problem in MD (ref), since protons can’t equilibrate freely.
For property prediction, an uneasy compromise can be reached wherein one takes a SMILES string, performs a conformational search, and then Boltzmann-averages properties over all different conformers, to return a set of values which are associated only with the SMILES string and not any individual conformation. (Matt Sigman does this, as does Bobby Paton for NMR prediction.) This is a lot of work, though.
“A Gulf Of Mutual Incomprehension”
These differences also become apparent when comparing the software packages that different tribes use. Take, for instance, the task of predicting partial charges for a given small molecule. A QM-tribe scientist would expect these charges to be a function of the geometry, whereas an MD-tribe scientist would want the results to be explicitly geometry-independent (e.g.) so that they can be used for subsequent MD simulations.
The assumptions implicit to these worldviews mean that it’s often quite difficult to go from QM-tribe software packages to MD-tribe software packages or vice versa. I’ve been stymied before by trying to get OpenMM and openforcefield to work on organic molecules for which I had a list of coordinates and not e.g. a SMILES string—although obviously coordinates will at some point be needed in the MD simulation, most workflows expect you to start from a topology and not an xyz file.
Similarly, it’s very difficult to get the graphics package NGLView to illustrate the process of bonds breaking and forming—NGLView is typically used for MD, and expects that the system’s topology will be defined at the start of the simulation and never changed. (There are kludgy workarounds, like defining a new object for every frame, but it’s nevertheless true that NGLView is not made for QM-tribe people.)
(I’m sure that MD-tribe people are very frustrated by QM software as well, but I don’t have as much experience going in this direction. In general, MD tooling seems quite a bit more advanced than QM-tribe tooling; most MD people I’ve talked to seem to interact with QM software as little as possible, and I can’t say I blame them.)
“A Curious Distorted Image of Each Other”
There are also cultural factors to consider here. The questions that QM-tribe scientists think about are different than those that MD-tribe scientists think about: a somewhat famous QM expert once told me that they were “stuck on an ivory tower where people hold their nose when it comes to DFT, forget anything more approximate,” whereas MD-tribe scientists often seem alarmingly unconcerned about forcefield error but are obsessed with proper sampling and simulation convergence.
It seems that most people have only a vague sense of what their congeners in the other tribe actually work on. I don’t think most QM-tribe scientists I know have ever run or analyzed a regular molecular dynamics simulation using e.g. AMBER or OpenMM, nor do I expect that most MD-tribe scientists have tried to find a transition state in Gaussian or ORCA. In theory, coursework could remedy this, but education for QM alone already seems chaotic and ad hoc—trying to cram in MD, statistical mechanics, etc is probably ill-advised at the present.
Social considerations also play a role. There’s limited crosstalk between the two fields, especially at the trainee level. How many QM people even know who Prayush Tiwary is, or Michael Shirts, or Jay Ponder? How many MD graduate students have heard of Frank Neese or Troy Van Voorhis? As always, generational talent manages to transcend narrow boundaries—but rank-and-file scientists would benefit immensely from increased contact with the other tribe.
“Unite Them”
I’m not an expert on the history of chemistry, but my understanding is that the two fields were not always so different: Martin Karplus, Arieh Warshel, and Bill Jorgensen, key figures in the development of modern MD, were also formidable quantum chemists. (If any famous chemists who read this blog care to share their thoughts on this history, please email me: you know who you are!)
And as the two fields advance, I think they will come closer together once more. As QM becomes capable of tackling larger and larger systems, QM-tribe scientists will be forced to deal with more and more complicated conformational landscapes: modern enantioselective catalysts routinely have hundreds of ground-state complexes to consider (ref), and QSimulate and Amgen recently reported full DFT calculations on protein–ligand complexes (ref).
Similarly, the increase in computing power means that many MD use cases (like FEP) are now limited not by insufficient sampling but by the poor energetics of the forcefields they employ. This is difficult to prove unequivocally, but I’ve heard this in interviews with industry folks, and there are certainly plenty of references complaining about poor forcefield accuracy (1, 2): a Psivant review dryly notes that “historically solvation energy errors on the order of 2–3 kcal/mol have been considered to be accurate,” which is hardly encouraging.
Many QM-tribe professors now work on dynamics: Dean Tantillo and Todd Martinez (who have long been voices “crying out in the wilderness” for dynamics) perhaps most prominently, but also Steven Lopez, Daniel Ess, Fernanda Duarte, Peng Liu, etc. And MD-tribe professors seem more and more interested in using ML mimics of QM to replace forcefields (e.g.), which will inevitably lead them down the speed–accuracy rabbit hole that is quantum chemistry. So it seems likely to me that the two fields will increasingly reunite, and that being a good 21st-century computational chemist will require competency in both areas.
If this is true, the conclusions for individual computational chemists are obvious: learn techniques outside your specialty, before you get forcibly dragged along by the current of scientific progress! There’s plenty to learn from the other culture of people that deals with more-or-less the same scientific problems you do, and no reason to wait.
As a denizen of quantum chemistry myself, I apologize for any misrepresentations or harmful stereotypes about practitioners of molecular dynamics, for whom I have only love and respect. I would be happy to hear any corrections over email.