If you’re wondering “is this a joke,” the answer is both yes and no.
This post is certainly a joke in the sense of "something that I'm intentionally doing because I thought it would be funny." I picked up Feser’s Scholastic Metaphysics: A Contemporary Introduction after reading a First Things article by Mary Harrington, and whilst reading the book I thought of the title of this post and started laughing. By analogy to Anselm’s ontological argument, the funniest version of this joke would have to be not just a title in my head but rather a full post published on my blog, and thus I was honor-bound to actually write the post, which you’re now reading.
Unfortunately, this post is also not a joke insofar as everything I’m writing is true: I actually read Feser’s Scholastic Metaphysics, I’m actually a B2B SaaS founder (yikes), and I’m actually going to write my sincere-yet-sarcastic thoughts about how the underappreciated field of Scholastic metaphysics connects to the scintillating topic of B2B SaaS.
Why write this post at all, though? Thomas Aquinas was skeptical of Anselm’s ontological argument, arguing that humans cannot sufficiently comprehend the divine, and one might be similarly skeptical of my claim that the full blog will be funnier than just imagining the title. B2B SaaS is a notoriously boring topic; it’s entirely possible that this joke just bogs down in philosophical terminology with few redeeming qualities.
My real goal here is to provide an interesting yet accessible review of the ideas in Feser’s book. Borrowing Scholastic language, one might say that while the efficient cause of this post is to entertain, the final cause (telos) of this post is to educate modern readers about concepts from medieval metaphysics. Metaphysics is a topic that can be particularly slippery for the modern secular–materialist worldview; while the world that we inhabit is certainly not free from metaphysical claims and ideas, metaphysics qua metaphysics is rarely discussed and remains a cultural blind spot.
Feser’s book does an excellent job pitching metaphysical ideas to the skeptical modern reader in accessible language, connecting these ideas to modern scientific notions of substance and matter. Accordingly, a serious blog-based book review in my usual style seems unlikely to provide differential value: I can hardly distill Feser’s already compact introduction any further without losing what little nuance and rigor remains. What follows is my attempt to instead blend sober-minded exploration of philosophy with low-effort B2B SaaS–related humor.
By way of quick background: metaphysics is the branch of philosophy concerned with the fundamental nature of reality, Scholasticism is the body of thought associated with late Medieval scholars like Thomas Aquinas, B2B refers to businesses selling to other businesses, and SaaS stands for “software as a service,” a business model wherein software is sold on an ongoing subscription basis.
I should also give a disclaimer that (1) I’m not a philosophy expert, just a guy who read a book, (2) these ideas are tough and I may be misunderstanding them, and thus (3) my summary of Feser’s writing is likely to be cartoonishly incorrect or simplistic to trained audiences.
Act and Potency
The first big idea of Feser’s book is the distinction between act and potency, immortalized in the first of the 24 Thomistic Theses endorsed by Pope Pius X in 1914 (p. 31):
Potency and act are a complete division of being. Hence, whatever is must be either pure act or a unit composed of potency and act as its primary and intrinsic principles.
These terms are slightly obscure. “Being-in-act” refers to an object as it really exists at present, while “being-in-potency” refers to what that object might become through change. The thesis’s claim is that, at core, the nature of things is such that the act–potency distinction is fundamental and prior to other differences (like difference in material, identity, and so on and so forth).
This description doesn’t replace or oppose conventional scientific descriptions of objects as comprising molecules, atoms, electrons, and so on. Rather, Feser argues that the metaphysical description of objects is orthogonal to and deeper than the reductionist materialist description, and that concepts of act and potency lose no value when applied to understanding e.g. the nature of subatomic particles. In fact, much contemporary philosophy-of-science discourse appears to be essentially recapitulating a potency-based description of scientific laws (see discussion on pp. 62–73), wherein the action of processes like gravitational attraction is mediated by causal relations which are themselves “real” and not purely happenstantial.
Feser argues that the proper relationship between act and potency provides the resolution to countless ancient philosophical quandaries. The Eleatic philosophers (Parmenides and Zeno) denied the reality of change, arguing that change implies that being must arise from non-being, which is impossible—hence Zeno’s well-known paradoxes. In parallel, Heraclitus argued that change alone describes reality and “there is no stability of persistence or even a temporary sort” (p. 33). Both of these views might seem ridiculous to us, and indeed can be shown to be self-inconsistent. (Feser adores using the “principle of retorsion” here, which refutes a position by showing it to be logically inconsistent with itself—I had to look it up.)
Instead, Feser argues that the correct view is to affirm both the reality of existence and change through the act–potency distinction (pp. 35–36, emphases original):
We have, then, the following basic argument for the distinction between potency and act: that change and permanence, multiplicity and unity, are all real features of the world cannot coherently be denied; but they can be real features of the world only if there is a distinction in things between what they are in act and what they are in potency.
How does this pertain to B2B SaaS? For software companies, being-in-act represents their current state—the product or services that they currently offer—while being-in-potency represents the products and markets they could conceivably address in the future.
Understanding the act–potency distinction is key for aspiring B2B SaaS companies. Startups that are pure act are unambitious, lacking a clear forward vision and any sense of change or industry disruption. A pure being-in-act SaaS company can be an excellent lifestyle business or PE-rollup target, but it’s unlikely to be a high-growth company or a “live player” in a fast-moving industry—and the lack of vision and ambition makes it hard to acquire and retain good talent and good customers.
In contrast, a pure being-in-potency SaaS business is unmoored from reality and exists without sufficient present substance to make good on its commitments. Such businesses tend to overpromise and struggle to deliver, often collapsing due to operational difficulties: Parker Conrad has some good reflections on the operational challenges faced at Zenefits as a result of too-rapid growth (see pp. 6–7 in this transcript from The Social Radars). In the worst-case scenario, pure potency businesses can become fraudulent, like Delve or Theranos.
Building a good B2B SaaS business, then, requires a keen understanding of the act–potency decision. The right business balances both act (what is) and potency (what might be), making sure to continue balancing these concepts as the company grows, employees turn over, and market conditions evolve.
Efficient Causes and Final Causes
Feser’s second chapter deals with the ideas of efficient cause and final cause (p. 88, emphases original):
Aristotelians famously distinguish between efficient and final causes. An efficient cause is that which brings something into existence or changes it in some way… It is, more or less, what is usually meant by “cause” in contemporary philosophy. A final cause is an end, goal, or purpose, “that for the sake of which” something exists or occurs.
This leads us to ideas like the principle of finality: “if A is by nature an efficient cause of B, then generating B must be the final cause of A” (92). This claim is controversial in modern philosophy, where efficient causes alone are typically seen to be sufficient to describe why an efficient cause necessitates its effects, but Feser argues that modern views ultimately collapse either to an Ockham-style divine occasionalism or an admission that there is something intrinsic about the relationship between causes and effects, some “intrinsic feature” of A that points it toward B (pp. 93–105). While final causes don’t provide a complete explanation of all causal relationships, they’re a useful and necessary component of the metaphysical landscape.
Here, the connection to B2B SaaS is obvious. Teleological statements, or statements pertaining to the “final cause” of a business, are shockingly common among Silicon Valley companies; take inter alia Airbnb’s goal “to create a world where anyone can belong anywhere,” Google’s aim to “organize the world’s information,” or Mark Zuckerberg’s statement that Facebook exists “to make the world more open and connected.”
The naïve metaphysical nihilist will be tempted to dismiss these statements as purely public relations or marketing, and to analyze these businesses solely on the basis of efficient causes: current cash-flow statements, quarterly earnings, and so on and so forth. This approach can work over short timescales, but struggles to make sense of high-variance strategic moves, like Zuck’s Metaverse conviction or ByteDance’s growing AI-for-science investments. Only teleological analysis based on the self-proclaimed final cause of these companies can explain such behaviors. (See also this CWT episode with Ben Thompson, where he argues that most founder-CEOs are indeed “true believers” and take their own grandiose mission statements seriously.)
Form and Matter
Aristotle famously outlines four causes for any effect: the formal cause, the material cause, the efficient cause, and the final cause. On this topic, Feser writes (p. 160, emphases original):
Our consideration of the theory of act and potency has led us to the latter two causes. A potency is always a potency for some actuality. It points beyond itself to an end or range of ends. Hence to understand a thing’s potencies is to understand it in terms of final causality. A potency can be actualized only by what is already actual. Hence to understand a thing’s coming into being or changing—that is to say, its becoming actual in various respects—is to understand it in terms of efficient causality. A thing’s final and efficient causes are extrinsic principles of its being, since the ends to which it points and the causes which actualize it are outside it.
Now the theory of act and potency also leads us naturally to two intrinsic principles of a thing’s being, namely its material and formal causes—that is to say, its matter and form.
Form refers to the “determinizing, actualizing pattern” that actualizes some potency, while matter refers to the “determinateable substratum that is the seat of the potencies in question” (p. 161). To illustrate this, Feser uses the example of ink inside a pen. The ink is matter that has the potency to dry into various shapes, like a circle or a star, and once dried has further potencies (like that of being erased). The ink is the matter, whereas the forms are the patterns like “being liquid,” “being circular,” “being blue,” and so forth; as the potencies of the ink are actualized, the forms are what result.
Feser next discusses the idea of “prime matter,” or matter that is completely devoid of form. While secondary forms of matter like ink are restricted in their potency (the ink cannot become a bottle of wine or a duck), prime matter “is not yet any particular thing or another… it is indeterminate, the pure potency for form” (p. 171). The modern scientific mind might think of atoms here, but even atoms have a metaphysical form (that of being atoms). Prime matter is so unformed that one cannot say anything about it other than that it is matter—it’s the raw substance from which all extant things are derived, through the action of various forms.
In the context of B2B SaaS, “prime matter” is obviously money. Money is pure potency to companies: through the action of another, it can become “secondary matter” in the form of employees, office space, software licenses, cloud credits, and so forth, but of its own accord money is “amorphous or without any form whatsoever” (p. 172). This too is an advantage, though—secondary matter is “limited to a particular time and place, and limited in the degree of perfection to which a thing instantiates them” (p. 171), but prime matter has no such limitations.
Cash in the bank can represent many things to B2B SaaS companies at various stages. Early on, it’s often a vanity metric: founders who raise more are successful, while small fundraises are shameful. Later on, cash becomes health, with distressed companies struggling to grow revenue, cut burn rate, and extend their runway long enough to hit their next fundraising milestones. But plenty of successful and cash-flow positive B2B SaaS companies continue to fundraise long after they can safely generate free cash flow from their existing business—why?
Reading Feser’s book helps me to understand why. Money is business’s prime matter, a direct infusion of potency “without form or void” which counteracts the natural accretive tendencies of successful institutions and preserves the key act–potency balance discussed above. And indeed companies which raise too much can suffer from the flaws of excessive potency discussed above, wasting resources on lavish office spaces and expensive conference booths rather than on substantial matters.
Substantial and Accidental Form
The Scholastics distinguished between substantial and accidental forms. An object with substantial form has a characteristic behavior “that derives from something intrinsic to it,” while an object with accidental form does not have such an intrinsic principle of behavior (p. 164). Trees naturally grow, seek light and water, and produce fruit, while a pile of rocks, however artfully arranged, does no such thing.
This dichotomy can be applied to businesses too. While a strict Scholastic would likely say that all companies have accidental forms, taking these terms loosely we can say that some companies have “substantial-ish forms” (i.e. forms which work together harmoniously towards a single goal) and other companies have “accidental-ish forms” (i.e. forms comprising disparate pieces with disparate goals). Companies with substantial-ish forms can naturally respond and adapt to their environment as a consequence of their organization and nature, while companies with accidental forms struggle to do so efficiently.
How can we determine which companies have substantial-ish forms, and how can we identify the ideal substantial form for a given company? Ronald Coase’s 1937 article “The Nature of the Firm” puts together an elegant model of the natural structure of companies under different constraints. Briefly, Coase argues that corporations emerge to minimize transaction costs. In a world without transaction costs, everyone could be an autonomous worker: developers could be hired ad hoc by their engineering managers, SDRs could be hired per lead, the CEO could pay the CFO for his services as needed, and there would be no companies. In the real world, though, the overhead associated with all of this would be tremendous—”hey, can I get you to sign another NDA for this next weekly meeting?”—and it’s simpler to create companies to minimize transaction costs within the organization. Thus a single B2B SaaS company that sells a discrete standalone product likely has a substantial corporate form.
Things start to get complicated, though, when contemplating ecosystem integration. Software conglomerates with few natural overlaps might be better off spinning out separate assets; many mergers destroy value, and it’s likely that the value of one overall conglomerate with accidental corporate form is lower than several divested entities with substantial corporate form. At the other extreme, some companies sell products that do not really deserve to be products qua products, but would make more sense as features of someone else’s product—if a merger would lower transaction costs throughout the system, both for vendors and customers, it might be possible that the proper substantial form is indeed the merged state. The current “death of SaaS” investor worries may stem from their belief that the substantial form of the frontier labs is, ultimately, to control all software.
Metaphysical Nihilists, Beware!
Feser argues that, despite the ignominity with which modern (i.e. post-1400s) philosophers treat Scholastic thought, the fundamental concepts of Scholastic metaphysics are nevertheless inescapable. Contemporary secular–materialist thought treats material and efficient causes as more or less sufficient, which leaves our culture dramatically ill-equipped to think critically about issues that cannot cleanly be reduced to an atomistic picture, including not only the nature of scientific laws but also of ideas, consciousness, and knowledge itself. On this topic Feser writes (pp. 27–28, emphases original):
The Scholastic maintains that there are truths of a metaphysical nature which (like the truths of logic and mathematics) are necessary and objective but which also (like the truths of logic and mathematics) are not plausibly regarded as propositions either of natural science or mere “conceptual analysis” [contra Hume]... This situation illustrates what is for the Scholastic a basic philosophical truth, which is that metaphysics is prior to epistemology. One way in which this is the case is that absolutely every epistemological theory rests on metaphysical assumptions—including Hume’s when he begins with the supposition that there are impressions and ideas, and including the naturalist’s when he supposes that our cognitive faculties are at least reliable enough to make natural science an objective enterprise…. When the critic of metaphysics insists that the metaphysician establish his epistemological credentials before making any metaphysical assertions, he is making a demand that is incoherent and to which he does not submit himself.
Feser’s argument is essentially that neglecting metaphysics is not an option, and that we’re all sneaking our metaphysics into our downstream philosophical or scientific thinking even if we don’t realize it. This isn’t, by itself, an argument that the Scholastic view is correct—but it should at least motivate the thoughtful modern reader to engage with Feser’s views and with metaphysics more broadly. Metaphysical nihilism is an unserious option.
Like Scholastic metaphysics, many modern observers (c. summer 2026) feel that B2B SaaS is dead and a thing of the past. Without belaboring the point too much—a full discussion would take an entirely separate post, or several—I’m very skeptical of these claims, for largely Coasian reasons, and think that most of the so-called “reinventions” of how software is sold, delivered, and maintained in a B2B environment are doomed to recapitulate much of the broad structure of classic mid-2010s B2B SaaS. (This blog post by Erik Bernhardsson of Modal is a great read and very relevant here.) If software is eating the world, B2B SaaS may be as inescapable as metaphysics.
There’s a lot more that one could imagine writing here—how the dispute between Duns Scotus and Aquinas on analogical equivocation mirrors different B2B SaaS deployment architectures, whether hylomorphism provides a good framework for understanding AI agent frameworks, what “software-enabled business transformation” really means in a metaphysical sense (is it the form of the business that’s changing?), the teleological implications of public benefit corporations—but every joke has to end.
If you liked this post, like and comment “PRINCIPLE OF SUFFICIENT REASON” below to get a free preview of my 12-week course “Supercharging Revenue Growth With the POWER of Scholastic Metaphysics.”
(n.b. please do not actually do this!)
Thanks to two philosophy professors (who shall remain nameless) for helpful discussions here, and Andrew White for sending me Erik's blog post.
TW: profanity, sarcasm, misuse of theological concepts.
Hosea and Gomer, from the Bible Historiale (1372)
There’s something special about “Venice Bitch,” the nine-minute-long song from Lana Del Rey’s 2019 album Norman Fucking Rockwell. Wikipedia writes that the song earned “unanimous praise from music critics” and “was ranked by numerous publications amongst the best songs of the year and decade,” while Tyler Cowen recently spoke about his affection for this song in a 2023 podcast with Rick Rubin, calling it his favorite song of recent years:
[“Venice Bitch”] just leaves me on the floor… It's one of those songs like “Strawberry Fields Forever” that it's even difficult to talk about, but it's a kind of dream pop, and it's creating a dream. It keeps on shifting like a dream. There's this extreme willingness to admit her passion for her lover's kiss, creating or harkening back to this world of an earlier America where things seemed much simpler, but the music is complex and it all comes together. And then there are seven more minutes of it where it becomes weirder. And you're always longing for the earlier, more melodic part to come back, and it never does, and it just makes you want to have to listen to it again.
The themes of “Venice Bitch” are, on the surface, obvious—nostalgia, lost love, memories of an idyllic time. But these ideas come from scattered images and, as Cowen notes, it’s hard to actually make sense of the song’s fragmented and metaphorical lyrics. Who is the lover? What happened to their relationship, why were they back together and then apart again, and what does the song’s fragmented and poetic ending mean? And, if nobody understands this song, why does it have such a peculiar hold over so many listeners?
Lana is no stranger to cryptic lyrics. In 2024, she told an interviewer that “I’ve put parts of my story into songs in ways that only I understand” and, when asked about her album’s story, replied “You have to figure it out.” While many writers have argued about the meaning of “Venice Bitch,” I think nobody’s yet unveiled the full esoteric meaning of this song. I will attempt to do so in this post.
More specifically, I’ll argue that behind the literal meaning of “Venice Bitch,” which refers to places and events in southern California, there’s a deeper allegorical/anagogical meaning to the song. Early church fathers, medieval authors, and Jewish mysticism all agree that there are multiple layers of meaning to any text, including not only the literal narrative but also moral and allegorical meanings designed to be understood upon deeper analysis and contemplation. As I’ll argue below, I think Lana Del Rey is part of this tradition and “Venice Bitch” cannot be fully understood solely through the surface-level narrative; instead, “Venice Bitch” is intended to poetically portray the futility of Temple-mediated propitiatory reconciliation in a fallen world.
(If you haven’t done so, it’s worth listening to the song before reading further.)
Expulsion from Eden
There’s little that’s concrete to start with in a song as meandering and poetic as “Venice Bitch.” One of the song’s only unambiguous references is the line “nothing gold can stay,” which comes from a 1923 Robert Frost poem. I’ll reproduce the poem here in its entirety, with emphases added:
Nature's first green is gold,
Her hardest hue to hold.
Her early leaf's a flower;
But only so an hour.
Then leaf subsides to leaf.
So Eden sank to grief,
So dawn goes down to day.
Nothing gold can stay.
Here, Frost uses the line “nothing gold can stay” to describe how the inherent beauty of creation always succumbs to decay in a post-Edenic world. The connection to Eden is particularly interesting for our purposes because in “Venice Bitch” the narrator writes about being “back in the garden” with her lover:
Back in the garden
We're getting high now because we're older
Me myself, I like diamonds
My baby, crimson and clover
If we entertain the idea that the “garden” might be Eden, what does it mean to be “back in the garden,” and how does this relate to “getting high now because we’re older”? Here we need to pull in another book from the Cowen Extended Universe, L. Michael Morales’s Who Shall Ascend the Mountain of the Lord? In this book, Morales argues that Eden is the original “holy mountain” of God, a lush garden-summit from which rivers flow to water the whole earth (pp. 51–52). In the “cultic cosmology” of the Pentateuch, ascent corresponds to movement towards God’s holy mountain while descent corresponds to movement away from God: hence why Abraham ascends Mount Moriah and Moses ascends Mount Sinai.
(It’s worth noting that the very first lines of “Venice Bitch” deliberately place us in a post-Edenic world: the narrator describes herself as dressing “in jeans and leather” in the first verse, echoing the “clothes of skins” from Genesis 3:21.)
Adam’s sin causes humanity to leave Eden and fall out of relationship with God, and even someone as righteous as Moses is unable to enter God’s presence by the end of Exodus. Within the narrative of the Pentateuch, only the Levitical temple cultus allows humans to once again reside in the presence of God and metaphorically “ascend the mountain of the Lord.” The tabernacle and later the temple thus serve as the new Eden, where God and man can dwell together—hence why the columns of the temple are carved to look like trees and hung with fruits. (G. K. Beale writes that “Israel’s earthly tabernacle and temple” were “reflections and recapitulations of the first temple in the Garden of Eden”; see The Temple and the Church’s Mission p. 66.)
So, through the mediation of the temple, we are allegorically “back in the garden” and “getting high” as we ascend to God's presence. This fits with the rest of the verse—”diamonds” reflects the high priestly breastplate covered in precious gems (which itself represents the entire cosmos, see Beale pp. 39–45), while “crimson and clover” refers to the blood-soaked hyssop used to paint the doorposts for Passover (Exodus 12:22) and, by extension, the idea that propitiatory sacrifice can appease God’s wrath.
(Why clover and not hyssop? Lana’s also alluding to the 1968 song “Crimson and Clover,” allowing the same line to serve two purposes.)
Exilic Longing
There’s more to “Venice Bitch” than a straightforward story of temple-mediated reconciliation, though. Let’s take one of the lines we discussed previously and zoom out to see the whole verse:
You're in the yard, I light the fire
And as the summer fades away
Nothing gold can stay
You write, I tour, we make it work
You're beautiful and I'm insane
The first line is simple enough: a reference to burnt offerings (“I light the fire”) conducted in the outer court of the temple (the “yard”). But here, the temple imagery is followed by the line that “nothing gold can stay,” suggesting that the imitation Eden of the temple is doomed just like the first Eden was and casting doubt on the whole temple enterprise.
What about summer fading away? In Song of Solomon, summer is associated with the presence of “the beloved” (Song. 2:8–13), which can be read literally as one’s lover but is also typically associated with God (what Kevin VanHoozer calls “an unbroken consensus for nearly nineteen centuries,” Mere Christian Hermeneutics p. 364). God is also represented by the sun in many other sections of the Bible (e.g. Malachi 4:2, Psalm 84:11). So the line “as the summer fades away” means that the presence of God is leaving the temple, recapitulating again the separation between God and man first experienced at Eden. God’s not leaving for good, but He’s growing more distant.
Since the remaining two lines of the verse occur after the decline of the temple cultus, it’s logical to associate them with the exilic and post-exilic prophets. The lyrics state that the narrator is touring—going around from place to place—while her lover is writing. This matches with imagery from Ezekiel, Hosea, and others which describe Israel as a faithless bride. While God writes through the prophets urging Israel to come back, Israel has abandoned their covenant and is wandering (“touring”) among the nations. Thus “you’re beautiful and I’m insane”; God’s love remains beautiful, but the narrator is not of sound mind and cannot accept this love (see also the opening refrain of “fear fun, fear love”).
The subsequent lines continue this theme:
Give me Hallmark
One dream, one life, one lover
Paint me happy in blue
What prevents this marriage from happening? Let’s jump right to the chorus:
Oh God, miss you on my lips
It's me your little Venice bitch
On the stoop with the neighborhood kids
Callin' out, bang bang, kiss kiss
Here, the narrator literally addresses God, saying she misses him “on my lips” (continuing the Biblical idea of the romantic love between God and his people, or possibly a Eucharistic reference). Unfortunately, she’s not with him—she’s “on the stoop with the neighborhood kids.” Morales writes that the Tower of Babel, most likely a ziggurat or ancient step pyramid, was an attempt to reconstruct the mountain of God from the city of man (pp. 61–62). By so doing, the nations attempted to take divine power into their own hands, recapitulating the sin of Eden and triggering divine retribution.
In these lyrics, the narrator is sitting on the steps with the neighborhood kids, making her a participant in the sin of Babel alongside the nearby nations. As she sits, she is making advances to passers-by. She’s not being faithful to her lover; instead, she’s been corrupted by the other nations and is flirting with idolatry. It’s with these same emotions that the hymnist Robert Robinson writes “Prone to wander, Lord, I feel it” and asks God to “bind my wandering heart to thee.”
Why is she a “Venice bitch”? The story of Hosea is pertinent here. In Hosea, God tells the prophet Hosea to marry a prostitute (Gomer) and make her his wife. Gomer is not faithful to Hosea and eventually leaves, and in chapter 3 Hosea has to go back and pay money to redeem her, either out of debt or slavery (the text isn’t quite clear). This story explains the narrator’s self-identification as a “Venice bitch”: put crudely, calling herself a “bitch” represents her unfaithfulness, while the inclusion of Venice makes it clear that she’s enslaved as well (before the Instagrammable bridges, Venice was principally known as a slave-trading empire).
The earlier mention of summer fading away can also be read as a reference to Israel’s faithlessness. Ezekiel 8 mentions the pagan practice of “weeping for Tammuz,” where worshippers would spend the hot summer months ritually mourning Tammuz, the sun- and spring-related deity who according to myth died and was reborn each year. According to Ezekiel, this ritual was sometimes performed in the temple itself (“in the yard”), demonstrating the depth of Israel’s wickedness and idolatry.
Captive to the Law of Sin
The images of unfaithfulness connect to the narrator’s early self-description as an “ice queen” who’s “trying to be stronger for you.” Her frozen heart echoes the prophetic image of “hearts of stone” (Ezekiel 36:26) which are unable to return God’s love properly; her futile attempts to be stronger echo Paul’s writing in Romans 7:
For I have the desire to do what is right, but not the ability to carry it out…. For I delight in the law of God, in my inner being, but I see in my members another law waging war against the law of my mind and making me captive to the law of sin that dwells in my members. Wretched man that I am! Who will deliver me from this body of death?
As Paul describes, the narrator is trapped in a cycle of sin and failure, her frozen and stony heart unable to respond as she ought to God’s love. While in her inner being she wishes for “one love,” her fallen nature makes this impossible, leading to a cycle of continued spiritual unfaithfulness and distance from the divine.
We can connect these ideas to a line from the song’s first verse: “live stream, I'm sweet for you.” Following the original streams of water coming out of Eden to water the earth, the idea of a “living stream” is a common Biblical image for God’s nourishment: Psalm 1 describes the man who follows God’s law as “planted by streams of water,” while Jesus offers the “living water” of the Spirit in John’s gospel (John 4:13–14, 7:37–39) and the Edenic temple-city at the end of Revelation contains “the river of the water of life, bright as crystal, flowing from the throne of God and of the Lamb” (Revelation 22:1). Much like the woman in John 4, the narrator identifies as “sweet for” (i.e. desiring) the “live stream” of the addressee, symbolizing her desire to return the presence of God—but, of her own power, she cannot obtain this living water.
Lana, Hermeneut?
At this point, we’ve been in the weeds long enough that the skeptical reader might wonder: would Lana Del Rey really write such a detailed and theological subtext to her song? I of course can’t answer this conclusively—as noted above, Lana writes complex and secretive lyrics with little explanations, making it difficult to know what she would or wouldn’t say.
Still, her Christian credentials are pretty strong:
Lana was raised Christian and has previously challenged Christian influencers that “I know the Bible verse for verse better than you do.” In a 2013 Nylon interview, she says that “I loved the mysticism, the idea of something bigger, the idea of a divine plan” (quoted in Relevant).
A 2023 Rolling Stone interview describes Lana’s experience of prayer: “you can pray and pray and pray to feel unburdened, and then sometimes, for no reason, on a Tuesday afternoon, everything lifts (quoted in Relevant).
She also featured pastor Judah Smith on her 2023 album Did You Know That There's a Tunnel Under Ocean Blvd. (Smith would go on to officiate her wedding in 2024.)
Deus Ex Machina
Let’s conclude by looking at the song’s ending. As Tyler notes, the song gradually shifts from a melodic opening to an increasingly atonal and experimental section in which the narrator repeats the lines “crimson and clover, honey” and “over and over, honey.” As mentioned above, “crimson and clover” reflects the propitiation of temple sacrifice. “Honey” represents the promise of God’s presence, a land “flowing with milk and honey” (Exodus 3:8, see also Judges 14:8–9 and 1 Samuel 14:24–30) while “over and over” shows us that the cycle of sin continues unabated, with no hope for improvement in the relationship between God and man. Even after the return from exile (“my baby’s back in town now”), the narrator pleads for the lover to return (“you should come, come over”) to no avail.
It’s against this chaotic backdrop that Lana suddenly sings the song’s final lyric, repeated five times:
If you weren't mine, I'd be jealous of your love
If you weren't mine, I'd be jealous of your love
If you weren't mine, I'd be jealous of your love
If you weren't mine, I'd be jealous of your love
If you weren't mine, I'd be jealous of your love
What does this abrupt and confusing ending mean? In Biblical language, jealousy is associated almost exclusively with God. God is a jealous God who wants his people’s love above all else (Exodus 20:5, 34:14; Deuteronomy 4:24, 5:9, 6:15). Here, it seems that the narrator has shifted and God himself has broken into the lyrics of the song. This literary device might seem unusual to modern audiences but it’s pretty common in the psalms: see, for instance, Psalm 12, Psalm 91, and Psalm 50.
God is responding to the endless cycle of “crimson and clover”–based attempts at reconciliation and asserting that He remains jealous for his people’s love. Just like in the prophets, God is promising to break the cycle of sin and create a new world in which His people can leave Him no more (cf. Jeremiah 31:34), achieving the promised mystical marriage between God and man: “one dream, one life, one lover” (cf. Revelation 21:2).
The song fades out here, leaving us uncertain how this can be accomplished. We can, of course, answer this question by referring to salvation history, the Gospels, or the New Testament. But we don’t even need to look that far to resolve this mystery. ”Venice Bitch” is a song by Lana Del Rey, which translated means “wool of the king.” What sort of king has wool? Only a king who is a lamb; namely, the Lamb Who Was Slain, who now sits enthroned in heaven (Revelation 5:6) and will one day bring justice and reconciliation to the world.
Something I’ve been thinking about recently is the information content of different biomolecules. While small molecules, peptides, antibodies, and oligonucleotides can all be valuable therapeutic assets in various contexts, they’re strikingly different to synthesize, develop, and simulate. There are well-known reasons for many of these differences—oligonucleotide synthesis can be highly automated, xenobiotic small-molecule metabolism proceeds through totally different pathways than peptide metabolism, and so on—but at a high level I think many of these differences can be seen as downstream of the observation that small molecules have much higher information entropy per atom.
Information entropy, also known as Shannon entropy (after Claude Shannon), quantifies the amount of “surprise” associated with each new piece of data. A sequence like “AAAAAAAAAAAAACAAAAA” has low entropy, since almost every letter is A—seeing another A gives us little new information, and so we can guess with pretty good odds that the next letter will be “A.” In contrast, a sequence like “ACTAGGACATAAGACAGGCT” has high entropy, since it seems that any position has four different possibilities. Since there are many possible sequences like this (just over a trillion for this length), each new letter conveys a lot of information about which particular sequence this is.
(This is a very brief introduction to Shannon entropy, and may be insufficient for those new to the topic—you can find plenty of better ones on Google.)
For molecules, we can approximate the information content per atom as the base-2 logarithm of the number of possible molecules divided by the number of possible atoms. This definition lets us make some quick estimates for the per-atom entropy of different modalities:
There are 4 valid nucleotides, or two bits of entropy per nucleotide. If we approximate a nucleotide as having 20 heavy atoms, we find that an oligonucleotide contains 0.1 bits of entropy per heavy atom.
For proteins and other peptides, there are 20 valid amino acids, or 4.32 bits of entropy per residue. Assuming 8.3 heavy atoms per residue, this gives us a value of 0.52 bits of entropy per heavy atom.
Small molecules are a different story. The GDB-17 paper estimates that there are 166 billion druglike molecules with 17 or fewer heavy atoms, with the vast majority of these having 15–17 heavy atoms. This corresponds to 2.2 bits of entropy per heavy atom.
The small-molecule value quoted above may even be conservative: GDB-17 applies fairly conservative filters and doesn’t include elements like S, P, B, and so on. If you take the oft-cited figure of 1060 possible drug-like molecules below 500 Da and approximate that as 35 heavy atoms, you arrive at a significantly larger value of 5.7 bits of entropy per heavy atom.
The markedly higher entropy of small molecules helps explain why small molecules are so tricky to synthesize. Fundamentally, any synthetic route must be specific and selective enough to disambiguate between virtually infinite numbers of potential products, which drives chemists to use complex and obscure reactions to achieve selectivity. Most approaches to simplifying small-molecule synthesis do so by vastly reducing the addressable space, enabling simple “Lego brick”–style routes to be employed. While there are sure to be improvements in synthetic technology over the decades to come, I think that making arbitrary small molecules will continue to be a difficult and complex task for fundamental and unescapable reasons.
The high information content of small molecules also explains why they can be such effective drugs. The ability to pack so much information into a small number of atoms makes it possible to achieve impressive selectivity with a tiny molecule—consider, e.g., the fact that you can have highly selective kinase inhibitors that are also small and non-polar enough to diffuse through the blood–brain-barrier. This sort of thing just isn’t possible with peptides!1
But the area where I’ve been thinking about this most is simulation and machine learning. It seems empirically true that it’s much easier to predict or model protein–protein binding than protein–small molecule binding. While protein-binder design with models like BindCraft works well and metrics like ipSAE seem to correlate well with protein–protein binding affinity, the analogous problems for small molecules still seem mostly unsolved (see e.g. Pat Walters’ writing from last year).
I think that this is downstream of information content. While a 300-residue protein has just as much total information as any small molecule, the overall complexity of any individual region of intermolecular interactions is much lower. There are a relatively small number of chemically distinct groups in proteins—indoles, imidazoles, amides, and so on—and it’s plausible that co-folding models or other biomolecular ML models can “learn” at a high level how these groups naturally interact with one another without needing to fundamentally understand the systems on the all-atom level. This means that learning to predict protein–protein or protein–oligonucleotide interactions is much easier than learning to predict protein–small molecule interactions, perhaps many orders of magnitude easier.
In contrast, there are almost infinitely many such small-molecule functional groups—pyridines, quinazolines, azaindoles, thiadiazoles, and so on—each with different chemical properties and a different interaction profile with protein sidechains. This means that the data scarcity problem is much worse than it seems for small molecules, and makes me skeptical that purely ML-based approaches for predicting binding affinity will work in the medium term. (I may be wrong here!)
(How much data will it take to actually learn arbitrary interatomic interactions? It’s hard to say for sure, but evidence from the neural-network-potential field suggests that it might take a lot. The OMol25 dataset comprises over 100 million DFT calculations with energy and per-atom force labels, so roughly 1–10 billion individual labels, and OMol25-trained models are the first models that seem to actually match the performance of physics-based methods on e.g. non-covalent interactions. While initiatives like OpenBind are promising and very valuable, I’m skeptical that even tens of thousands of new protein–ligand complexes will be enough here.)2
I remain optimistic about the future of physics and physics-adjacent methods in small-molecule drug design for these reasons. Methods like quantum chemistry and FEP are able to avoid the training-data limitations of pure ML methods and show good generalizability for arbitrary small molecules. While I’m unbelievably excited about our new AI-powered scientific future, I think that the immense information content of small molecules puts fundamental limitations on what ML can accomplish, and means that (for better or worse) we’re going to be stuck with physics for the foreseeable future.
Thanks to Ishaan Ganti and Ari Wagen for helpful discussions here.
Footnotes
Except for some peptides and other large molecules, which do diffuse through the blood–brain barrier! Some of these seem to occur through active transport, but there’s some mystery here still.
Note that there are many different potential forms of data, the shape of these problems is different, and there are other issues that make this comparison imperfect. I use this analogy simply to argue that we probably need a lot more data, not a little.
June 18, 2026The Rebuilding of the Temple is Begun, Gustave Doré (1866)
As AI systems get more powerful and better at scientific tasks, what will the new research ecosystem look like?
One perspective, which is rarely stated explicitly but comes up implicitly or in conversation, is that AIs will render much of conventional theory and modeling obsolete: either AI will write its own scientific tools and simulations, or AI will somehow reason from first principles and circumvent the need for conventional simulation tools at all. This view often presumes that conventional approaches to understand or model natural phenomena are flawed or incapable and that LLMs are better off ditching the old ways & starting from scratch.
As someone who works full-time on simulation tools for chemistry and materials science, I’m clearly a bit biased against this take. But I think I’m not alone here; the vast majority of actual industry scientists I know who are optimistic about AI are eager to give their agents access to the full panoply of tools that they themselves rely on.
In this post I want to briefly defend the thesis that we should be excited about the prospect of giving design + simulation tools to scientific AI agents. I think that there are real durable advantages to tool integration. Rather than feeling like agentic tool use is a short-term crutch until the true “bitter lesson” kicks in, I believe that we should expect scientific agents to use tools indefinitely, and I expect tool integration to be an increasingly important aspect of frontier agentic science.
(Long-time blog readers may recall that I first wrote about these ideas in my October 2025 posts about “AI scientists”, scare quotes deliberate. Since writing that post, I’ve been spending an increasing fraction of my time working with both AI agents inside Rowan and external companies building AI agents atop Rowan. This post attempts to distill some of the ways that I feel my thoughts have changed or sharpened since then.)
In my mind, there are four big advantages to giving LLMs access to dedicated scientific tools.
1. Tools As Domain-Specific Crystallized Intelligence
Ezra Reads the Law to the People, Gustave Doré (1866)
In psychology, fluid intelligence is the raw ability that an individual has to flexibly solve totally new problems, while crystallized intelligence is their ability to use existing knowledge and expertise to solve problems similar to those they’ve encountered before (see this Wikipedia article for more details, nuance, and sources). Crystallized intelligence is what differentiates experts in a field from newcomers. While working on frontier science problems for years probably won’t increase your fluid intelligence, it’ll certainly increase your crystallized intelligence for similar problems.
Specialized scientific tools can’t increase the raw fluid intelligence of a model, but they can act as reservoirs of crystallized intelligence to deploy against specific problems. Giving LLMs access to scientific tools (and skills) is a simple way to inject a certain amount of expertise into the conversation in a token- and context-efficient way.
To make this concrete, let’s think about a fairly simple task: running a MD simulation starting from a PDB structure of a protein–ligand complex. To do this from scratch, an agent has to figure out how to solve lots of moderately tricky sub-problems, To name just a few:
How should the protein structure be prepared?
Does the protein have hydrogens? If not, how should they be added?
Are there unresolved residues?
Are there any crystallization artifacts that need to be removed?
What forcefield should be chosen for the protein, the ligand, and the water?
And how should the ligand be parameterized? (Is there a starting topology?)
How should the system be simulated?
What thermodynamic ensemble?
What forcefield settings, barostat, thermostat, and so on?
How long should equilibration run for?
How many replicates should be run?
What properties should be analyzed? What insights are we looking for, and how do we rigorously extract these without introducing bias?
Modern LLMs are quite competent at problems like this and can often figure out good answers to all of these questions, if given time and the ability to iterate & self-correct. The models have read lots of papers and have substantial latent expertise. But, just like a person doing a new task for the first time, LLMs often spend a substantial amount of time, tokens, and context experimenting with various parameters and trying to figure out the best way to approach a given problem.
This is fine in isolation but quickly becomes a drag in the process of larger projects. If you’re trying to get an LLM to run a complex scientific process autonomously (i.e. “screen these compounds with docking, run MD, identify the important interactions, score them with SAPT, and propose 20 new compounds”), then the LLM needs to be able to confidently and routinely run each individual step to be able to execute the entire end-to-end workflow. Providing an LLM with purpose-built tools/APIs that work out of the box is a simple way to accomplish this.
This is already how tools like Claude Code and Codex work so well. Rather than reinventing version control, filesystems, sandboxes, and subprocesses from first principles, coding agents excel because they’re able to leverage pre-existing high-level tools like Git and focus only on the important and differentiated tasks.
2. Tools As World Model
Another advantage of simulation tools is that they act as an external check on LLMs’ internal reasoning. This is particularly salient for many problems in chemistry; reasoning about 3D structures without a visual aid is notoriously difficult, which is why organic chemistry is such a visual discipline and why ball-and-stick molecular modeling kits have been a mainstay of chemistry education for decades.
While AI agents are generally very smart, they still mostly operate based on text-based representations and can struggle to reason correctly about 3D problems.* Starting from just a PDB file and a SMILES string, it’s not very easy to tell if a given molecule will fit into the pocket or which residues it’s likely to interact with: expert human chemists aren’t good at this task, and even a superintelligent agent is unlikely to prefer interacting with chemistry in this way.
Using a tool like docking or co-folding here is just a better choice for the problem—it’s built to handle this input modality, works reproducibly, and serves as an external check on LLM reasoning. The agent might really think that a given molecule should fit into the ATP binding site, but actually running the docking calculation will tell the agent whether or not this is true.**
In practice, I think this sort of external “sanity check” helps to keep agents on track and prevent them from hallucinating too much. It can be dangerous for “AI scientists” to think for too long without testing their ideas, much as it is for human scientists. Simulation tools provide a simple and fast way to check ideas before committing to costly or time-consuming laboratory testing.
This is analogous to how static type checking and other automated code-review tools have become so important in the age of agentic coding (as my colleague Jonathon’s written about). Even frontier LLMs still make trivial errors when writing code, but simple easy-to-run sanity checks that exist outside the LLM’s control can dramatically reduce the error rate and increase the quality of the resultant code.
* I think this is one of the reasons why math and programming have seen so much more dramatic progress than chemistry, biology, and materials science.
** Docking is not a good tool for predicting binding affinity, but it’s pretty good at predicting if something’s too big to fit in the pocket.
3. Tools As Efficient Test-Time Compute
Large language models are, as the name implies, large and expensive to run. Even if one assumes that the LLMs of the future possess superhuman reasoning powers and (following the above example) can dock compounds using only string representations of protein and ligand, it’s very unlikely that this will be a pragmatically useful way to run these calculations.
Math is a helpful case study here. I can do arithmetic up to some scale in my head, but pretty soon I prefer to use a calculator (particularly if I want to ensure I get the answer right). Similarly, LLMs are perfectly capable of doing math on their own, but ask them to compute a tricky trigonometric equation and they far prefer to open a Python notebook. Computers are notoriously good at doing math, and it’s much more efficient to do math directly on a computer than to essentially emulate a calculator in the weights of an LLM.
I have similar feelings about asking LLMs to do complicated chemical prediction problems without tools. While it’s great to see that Claude is able to predict NMR shifts with accuracy similar to (or even exceeding) that of ChemDraw / MNova,* I personally would far prefer a physics- or ML-based NMR prediction model that runs quickly and can be systematically improved with more data.** Quantitatively accurate NMR prediction is just not a task that Claude needs to excel at! It would be much more efficient to give Claude an NMR tool and save its context window for the difficult-to-encode parts of the task (like spectrum-to-structure inductive reasoning).
The cost and token savings of using simple simulation tools instead of frontier LLMs will matter too. As agentic science moves beyond demos and towards wider adoption, workflow cost will become more important (as is already happening for big engineering teams), and asking Fable or GPT 6 to do routine cheminformatics or property-prediction tasks will be not only inefficient but impractically expensive. The right scientific tool can serve as a much more efficient form of test-time compute than naive LLM reasoning.
* While this is a bit of a tangent, I would also note that the ChemDraw/MNova NMR-prediction tools are hardly state-of-the-art; I’m pretty sure that modern spectrum-prediction models will handily outperform Claude here.
** Early readers note that Claude too can be improved by looking at more NMR data. While this is true, it’s a bit harder to fine-tune an LLM than simply refitting a ChemProp model like many pharma teams do weekly; maybe this will change in the future.
4. Tool Calls As Audit Log
GPT Image 2–generated illustration of the angel with the writing case from Ezekiel 9, in the style of Gustave Dore.
While we may one day be comfortable letting our “AI scientists” run rogue and queue whatever lab experiments they want, in the short term I suspect that most organizations would prefer to have an idea of what their agents are doing and why they’re making a given decision. Much as in coding, external tool calls can serve as an interpretable signal about what an LLM is doing.
Companies following a predict-first paradigm (inter aliaBMS) already routinely run all their human-designed molecules through the same battery of simulations; asking an LLM to use the same tools makes it easy to compare an agent’s results and reasoning to what humans are already doing. In cases where agents are working well, this will likely increase institutional trust; if they start to perform poorly, the external audit log of tool calls might be a helpful tool in figuring out what went wrong.
Fundamentally, tool calls are also a nicely interpretable way to track LLM decision-making. Scientists already have a fundamental understanding of what different scientific tools are useful for; seeing an agent run a torsion scan or a SAPT calculation between a ligand and a given protein residue is an easy way for scientists to quickly grok what a LLM is doing, right or wrong.
There’s certainly additional nuance that’s missed by the above reasons, but I think this captures the spirit of my objections to the “no tools” position. In other domains—software development, math, CS, and so on—it seems like LLMs are increasing the need for high-quality tooling, not decreasing it, and I think we should expect the same trend to continue in science.
Thanks to Ari Wagen, Eli Mann, Eugene Kwan, and Ishaan Ganti for helpful comments on this post, and to many others for conversations on these topics.
Yesterday, an interesting paper was released by Prescient Design (Genentech), “Evaluating the Progression of Large Language Model Capabilities for Small-Molecule Drug Design.” The authors formulate a variety of problems in small-molecule drug design as reinforcement-learning (RL) problems and use RL-based post-training to improve the performance of a 30B-parameter Qwen model (specifically, Qwen3-30B-A3B-Thinking-2507). They then compare the performance of this fine-tuned Qwen model (“Aspen”) to various frontier models on property prediction, molecular representation tasks, and property-constrained generation.
I was particularly interested in the authors’ demonstration that both Aspen and frontier models like GPT 5.2 & Claude Opus 4.6 could perform well in simulated lead-optimization scenarios. Here’s how the authors describe this evaluation in the paper (newlines added for visual clarity):
Next, we examine the capabilities of these models in a multi-turn setting, consisting of an iterative molecular optimization loop that simulates real-world lead optimization. Specifically, we consider optimizing a docking score—as a weak proxy for potency—under a set of property constraints, consisting of a combination of DMPK and RDKit properties that can all be computed.
For the experiments here, we consider a single target, a carbonic anhydrase IX (PDB ID: 8TTR), and a starting seed molecule obtained from (Elshamsy et al., 2025). Each trajectory starts from the same molecule and runs for 20 turns; at each turn the model proposes a modified SMILES and is provided with the resulting docking score and the corresponding property values of the constraints (see Appendix C.2). In the system prompt (see Appendix C.1), we instruct the model to always propose structurally novel molecules across all 20 turns.
Importantly, we blind the target name from the model, resulting in a black-box optimization setting that restricts the model from leveraging knowledge about the protein to carry out the optimization.
This sort of multiparameter optimization is a relatively non-trivial task; while it’s relatively easier to increase docking scores by simply making the molecules bigger or adding hydrogen-bond donors, it’s substantially harder to generate useful improvements that don’t simultaneously compromise solubility, permeability, and other relevant drug-like properties. As such, it’s standard to use specialized frameworks like REINVENT for tasks like this (and even these tools have well-known pathologies).
What the Genentech team did is much simpler. Essentially, they:
Tell an LLM that it’s supposed to act like a medicinal chemist, what the design target is, and what the initial SMILES is.
Let it propose a new SMILES.
Score the SMILES using various computational methods.
Return the results to the LLM and let it try again.
Surprisingly, this works! Over time, the LLM-proposed molecules get better (as assessed by docking score) while generally abiding by the requested constraints. Although the guess-and-check nature of this workflow implies that even random perturbations should eventually lead to improvements, several factors suggest that chemical intelligence is actually at play here: (1) the base Qwen model is barely able to improve over the baseline, suggesting that it’s unable to meaningfully reason about molecule structures, and (2) the later models from each family fare better than the earlier models, suggesting that increased intelligence leads to better optimizations even within a model family.
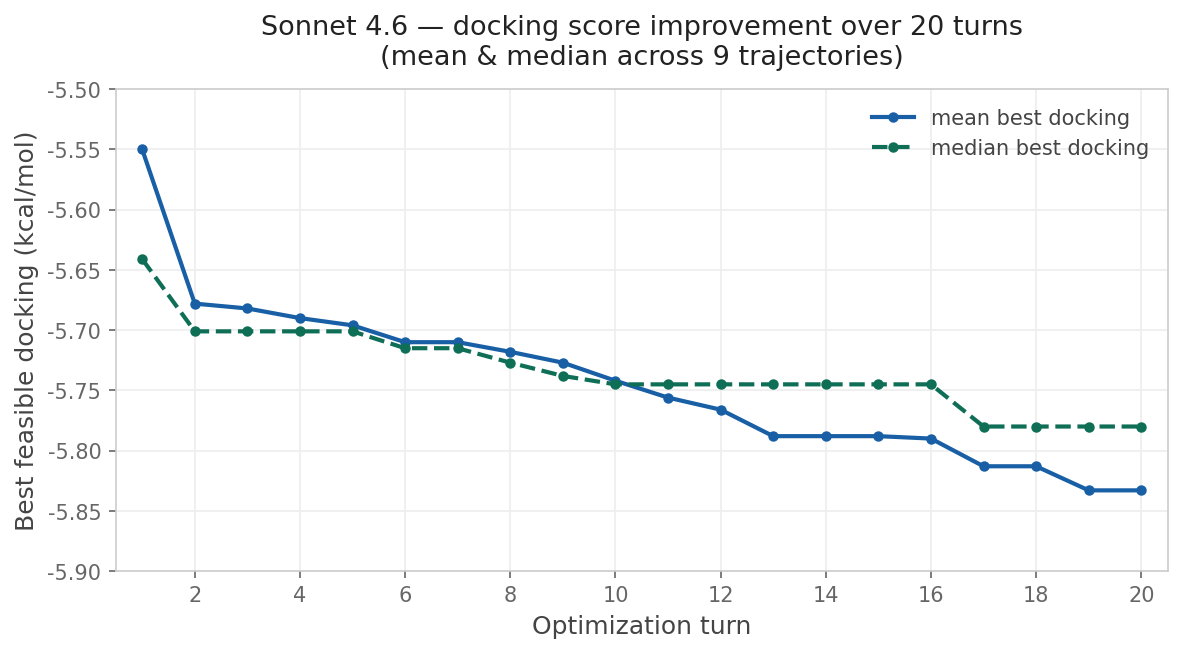
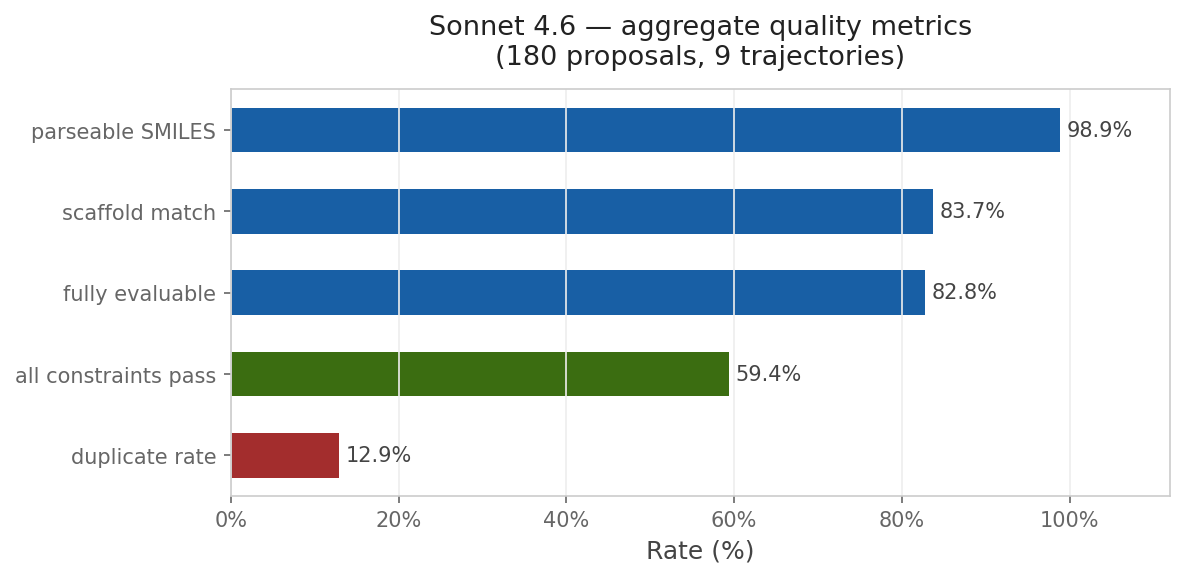
This approach is compelling because of its simplicity. To run the reported multi-parameter optimization approach, you don’t need a fancy pre-trained generative model or a library of potential synthons: instead, all you need is your favorite LLM and a way to score candidate molecules. I wanted to see if I could recapitulate this with Rowan, so I used Claude Code to build an optimization loop around Rowan’s API. I copied the system prompt almost exactly from Genentech and used Rowan’s docking, solubility, and membrane-permeability workflows to score each candidate compound. While the specific numbers are different than those reported by the Genentech team, the trend is clear:
Although this particular demonstration (optimizing a docking score on carbonic anhydrase) is unlikely to be incredibly impactful, the approach is incredibly general—any computational oracle that can be run via API can, in theory, be optimized using this approach. One could imagine optimizing kinome selectivity, tuning free-radical stability in a polymerization inhibitor, designing molecules with desired optical properties, or any number of other applications.
And while the present implementation is relatively light on scientific context, even blinding the protein’s identity so that the LLMs can’t cheat with pre-existing carbonic anhydrase knowledge, it’s not hard to imagine using data from previous projects or existing scientific literature to help the LLM devise new ideas. (Even providing a representation of the protein–ligand binding site would probably be very helpful.) There’s a lot of ways that this can be improved!
Above that, we can imagine “AI scientists” managing well-defined multi-parameter optimization campaigns. These agents can work to combine or orchestrate the underlying simulation tasks in pursuit of a well-defined goal, like a given target–product profile, while generating new candidates based on scientific intuition, previous data, and potentially human input. Importantly, the success or failure of these agents can objectively be assessed by tracking how various metrics change over time, making it easy for humans to supervise and verify the correctness of the results. Demos of agents like this are already here, but I think we’ll start to see these being improved and used more widely within the next few years.
This is exactly what we’re seeing now. Despite having been optimistic about agentic AI–driven optimization for months now, I’m still impressed that a single evening of Claude Code (plus Rowan’s API) can produce a competent med-chem optimization agent—no domain-specific fine-tuning or specialized literature access necessary. Capabilities are advancing incredibly quickly in this field, and it’s possible to do things today that were hard to imagine a year ago.
I’ve put all my code in a GitHub repository; if you’re interested in this approach, feel free to clone the repository and modify it however you see fit (oracles, system prompt, LLMs, etc). Full methodological details are in the repo.