Since my previous “based and red pilled” post seems to have struck a nerve, I figured I should address some common objections people are raising.
Although this is obvious, I wanted to preface all of this by saying: this is my opinion, I'm not some expert on systems of science,
and many of the criticisms come from people with much more scientific and institutional expertise than me.
It's very possible that I'm just totally wrong here!
But what I'm saying makes sense to me, and (it seems) to a lot of other people, so I think it's at least worth having this discussion.
Commenters Who Feel 13C NMR Is Scientifically Crucial
A few people pointed out that there are lots of instances in which carbon NMR is very important
(1,
2,
3,
4,
5).
I don't disagree with this at all; I've also used 13C NMR to solve problems that 1H NMR and mass spectrometry alone couldn't solve!
But just because it’s crucial sometimes doesn’t mean it’s crucial all the time.
Does anyone really think that you need carbon NMR to tell if Boc protection of a primary amine worked?
Most of the reactions that people do—especially people for whom synthetic chemistry is a means and not an end—are pretty straightforward, such that I think it’s fair to assume you could deduce the correct product with high confidence without 13C NMR.
(Again, if carbon spectra were so crucial, it wouldn’t be the case that many people don’t get them until the very end of the project.)
Commenters Who Feel That It's Important To Have Non-Crucial Data To Test Your Hypotheses
This point was also made by a number of people
(1,
2,
3),
perhaps most succinctly by “Chris Farley”:
Never meet your heroes.
I think this is an important point—part of what we ought to do, as a scientific community, is challenge one another to test our hypotheses and scrutinize our assumptions. Nevertheless, I’m not convinced this is a particularly strong argument for carbon NMR specifically. What makes 13C{1H} spectra uniquely powerful at challenging one’s assumptions, as opposed to other data?
Keith Fritzsching points out that HSQC is much faster and gives pretty comparable information (as did other readers, privately), and simply replacing 13C with HSQC in most cases seems like it would nicely balance hypothesis testing with efficiency.
(Relatedly, Xiao Xiao recounts how reviewers will request 13C spectra even when there’s plenty of other data, including HSQC and HMBC. This is a pretty nice illustration of how powerful status quo bias can be.)
Commenters Who Say Carbon Spectra Are Easy To Acquire
(cue flashbacks to undergraduate me trying to dissolve enough of some tetracyclic monster in pyridine-d5 to see my last quat)
I've heard this from plenty of people before, and it's true that sometimes it's not hard at all to get a nice carbon spectrum! But sometimes it is really hard, also.
Based on the other responses, it seems like lots of other people agree with this sentiment.
(Is it possible that some of this disagreement reflects whether one has access to a helium cryoprobe?)
Commenters Who Feel It's Important To Have Consistent Journal Standards
A few people pointed out that making carbon NMR necessary on a case-by-case basis would be burdensome for editors and reviewers, since they'd have to think through each case themselves
(1, 2).
This is a fair point, and one I don't have a good response to.
However, it's worth noting that this is already what we do for pretty much every other claim, including many complex structural problems: give the data, draw a conclusion, and ask reviewers to evaluate the logic.
Arguments about where the burden of proof should lie are tedious and usually unproductive, but I think we should have a pretty high barrier to making specific methods de jure required for publication.
Commenters Who Dislike My Claim That Journals Could Permit More Errors
I'm going to highlight Dan Singleton here, someone I respect a ton:
The thread goes on, obviously, and is worth reading.
I’m not trying to suggest that journals ought not to care about accuracy at all; ceteris paribus, accuracy should be prioritized. But given that we’re all constrained by finite resources, it’s important to consider the tradeoffs we’re making with every policy decision. It’s possible that trying to increase accuracy by asking for more data could have deleterious consequences:
There’s clear extremes on both ends: requiring 1H NMR spectra for publication is probably good, but requiring a crystal structure of every compound would be ridiculous.
I think it’s easiest to think about these issues in terms of two separate questions: (1) relative to where we are today, should we push for more accuracy in the literature or less, and (2) are we achieving our chosen degree of accuracy in the most efficient manner possible?
The first question is clearly complex, and probably deserves a longer and fuller treatment that I can provide here—although I’ll note that others have espousedmore radical positions than mine on peer review (h/t Michael Bogdos for the second link). I hope to write more on this subject later.
But the second question seems more straightforward. Is requiring 13C NMR for every compound a Pareto-optimal way to ensure accuracy, as opposed to HSQC or HMBC? I struggle to see how the answer can be yes.
13C NMR is, generally speaking, a huge waste of time.
This isn’t meant to be an attack on carbon NMR as a scientific tool; it’s an excellent technique, and gives structural information that no other methods can. Rather, I take issue with the requirement that the identity of every published compound be verified with a 13C NMR spectrum.
Very few 13C NMR experiments yield unanticipated results. While in some cases 13C NMR is the only reliable way to characterize a molecule, in most cases the structural assignment is obvious from 1H NMR, and any ambiguity can be resolved with high-resolution mass spectrometry. Most structure determination is boring. Elucidating the structure of bizarre secondary metabolites from sea sponges takes 13C NMR; figuring out if your amide coupling or click reaction worked does not.
The irrelevance of 13C NMR can be shown via the doctrine of revealed preference: most carbon spectra are acquired only for publication, indicating that researchers are confident in their structural assignments without 13C NMR. It’s not uncommon for the entire project to be scientifically “done” before any 13C NMR spectra are acquired. In most fields, people treat 13C NMR like a nuisance, not a scientific tool—the areas where this isn’t true, like total synthesis or sugar chemistry, are the areas where 13C NMR is actually useful.
Requiring 13C NMR for publication isn’t costless. The low natural abundance of 13C and poor gyromagnetic ratio means that 13C NMR spectra are orders of magnitude more difficult to obtain than 1H NMR spectra. As a result, a large fraction of instrument time in any chemistry department is usually dedicated to churning out 13C NMR spectra for publication, while people with actual scientific problems are kept waiting. 13C NMR-induced demand for NMR time means departments have to buy more instruments, hire more staff, and use more resources; the costs trickle down.
And it’s not like eliminating the requirement to provide 13C NMR spectra would totally upend the way we do chemical research. Most of our field’s history, including some of our greatest achievements, were done in the age before carbon NMR—the first 13C NMR study of organic molecules was done by Lauterbur in 1957, and it would take even longer for the techniques to advance to the point where non-specialists could use the technique routinely. Even in the early 2000s you can find JACS papers without 13C NMR spectra in the SI, indicating that it's possible to do high-quality research without it.
Why, then, do we require 13C NMR today? I think it stems from a misguided belief that scientific journals should be the ultimate arbiters of truth—that what’s reported in a journal ought to be trustworthy and above reproach. We hope that by requiring enough data, we can force scientists to do their science properly, and ferret out bad actors along the way. (Perhaps the clearest example of this mindset is JOC & Org. Lett., who maintain an ever-growing list of standards for chemical data aimed at requiring all work to be high quality.) Our impulse to require more and more data flows from our desire to make science an institution, a vast repository of knowledge equipped to combat the legions of misinformation.
But this hasn’t always been the role of science. Geoff Anders, writing for Palladium, describes how modern science began as an explicitly anti-authoritative enterprise:
Boyle maintained that it was possible to base all knowledge of nature on personal observation, thereby eliminating a reliance on the authority of others. He further proposed that if there were differences of opinion, they could be resolved by experiments which would yield observations confirming or denying those opinions. The idea that one would rely on one’s own observations rather than those of others was enshrined in the motto of the Royal Society—nullius in verba.
nullius in verba translates to “take no one’s word for it,” not exactly a ringing endorsement of science as institutional authority. This same theme can be found in more recent history. Melinda Baldwin’s Making Nature recounts how peer review—a now-core part of scientific publishing—became commonplace at Nature only in the 1970s. In the 1990s it was still common to publish organic chemistry papers without supporting information.
The Royal Society—a group of folks who definitely didn't take 13C NMR spectra.
The point I’m trying to make is not that peer review is bad, or that scientific authority is bad, but that the goal of enforcing accuracy in the scientific literature is a new one, and perhaps harder to achieve than we think. There are problems in the scientific literature everywhere, big and small. John Ioannidis memorably claimed that “most published findings are false,” and while chemistry may not suffer from the same issues as the social sciences, we have our own problems.
Elemental analysis doesn’t work, integration grids cause problems, and even reactions from famous labs can’t be replicated. Based on this, we might conclude that we’re very, very far from making science a robust repository of truth.
Nevertheless, progress marches on. A few misassigned compounds here and there don’t cause too many problems, any more than a faulty elemental analysis report or a sketchy DFT study. Scientific research itself has mechanisms for error correction: anyone who’s ever tried to reproduce a reaction has engaged in one such mechanism. Robust reactions get used, cited, and amplified, while reactions that never work slowly fade into obscurity. Indeed, despite all of the above failures, we’re living through a golden age for our field.
Given that we will never be able to eradicate bad data completely, the normative question then becomes “how hard should we try?” In an age of declining research productivity, we should be mindful not only of the dangers of low standards (proliferation of bad work) but also of the dangers of high standards (making projects take way longer). There’s clear extremes on both ends: requiring 1H NMR spectra for publication is probably good, but requiring a crystal structure of every compound would be ridiculous. The claim I hope to make here is that requiring 13C NMR for every compound does more to slow down good work than it does to prevent bad work, and thus should be abandoned.
One of the more thought-provoking pieces I read last year was Alex Danco’s post “Why the Canadian Tech Scene Doesn’t Work,” which dissects the structural and institutional factors that make Silicon Valley so much more effective at spawning successful companies than Toronto.
I’ll briefly summarize the piece’s key arguments here, connect it to some ideas from Zero to One, and finish by drawing some conclusions for academia.
Danco’s key insight is applying James Carse’s distinction between finite and infinite games to entrepreneurship. What makes a game “finite” or “infinite”?
First, finite games are played for the purpose of winning. Whenever you’re engaging in an activity that’s definite, bounded, and where the game can be completed by mutual agreement of all the players, then that’s a finite game. Much of human activity is described in finite game metaphors: wars, politics, sports, whatever. When you’re playing finite games, each action you take is directed towards a pre-established goal, which is to win.
In contrast, infinite games are played for the purpose of continuing to play. You do not “win” infinite games; these are activities like learning, culture, community, or any exploration with no defined set of rules nor any pre-agreed-upon conditions for completion. The point of playing is to bring new players into the game, so they can play too. You never “win”, the play just gets more and more rewarding.
In entrepreneurship, Danco argues that infinite games are good and finite games are bad. Good founders are playing infinite games: they want to build something important and keep on contributing to society and progress. In contrast, bad founders are playing to win a finite game—acquiring lots of funding, getting high valuation, or exiting with a big IPO.
Danco identifies numerous ways that the Canadian startup ecosystem incentivizes finite games at the expense of infinite games. One important factor favoring finitude is the scrutiny given to deals between founders and funders (e.g. in a seed round), which tends to favor conservative or incremental ventures over ambitious, idealistic ones:
[High deal scrutiny] is bad, for two reasons. It’s bad because the very best startups, who have the longest time horizon and are most curious about the world, will look disproportionately uninspiring. They’ll have the fewest definite wins relative to their ambition, and the most things that can potentially go wrong.…
Conversely, [high deal scrutiny is] bad because startups will learn to optimize for how to get funded. So if seed deals take 3 months, then founders will learn to build companies that look good under that kind of microscope. And that means they’re going to optimize for playing determinate games, so that they can show definable wins that can’t be argued against; rather than what they should be focusing on, which is open-ended growth.
Government support for startups also tends to prioritize finite games at the expense of infinite games, since the requisite bureaucracy tends to stifle fast-moving innovation:
The problem with [research tax] credits, honestly through no fault of their own, is that you have to say what you’re doing with them. This seems like a pretty benign requirement; and honestly it’s pretty fair that a government program for giving out money should be allowed to ask what the money’s being used for. But in practice, once you take this money and you start filling out time sheets and documenting how your engineers are spending their day, and writing summaries of what kind of R&D value you’re creating, you are well down the path to destroying your startup and killing what makes it work.
Overall, Danco paints a picture of a place where an obsession with goals and benchmarks has almost completely crowded out sincere innovation:
Quite in character with our love of milestones, Canada loves anything with structure: accelerators, incubators, mentorship programs; anything that looks like an “entrepreneurship certificate”, we can’t get enough of it. We’re utterly addicted with trying to break down the problem of growing startups into bite-size chunks, thoughtfully defining what those chunks are, running a bunch of promising startups through them, and then coming out perplexed when it doesn’t seem to work.(emphasis added)
While Danco limits himself to comparing Canada and Silicon Valley, this failure mode is sadly not confined to Canada. Indeed, many of his observations are directly applicable to academic research. The large number of finite games in academia—publishing papers, writing a dissertation, submitting grants, getting tenure—tends to crowd out the more impactful infinite games that lead to real, meaningful progress, and promotes incremental projects with a high chance of success. (This is simply Charles Hummel’s “tyranny of the urgent” by a different name.)
My claim is that the distinction between finite and infinite games is best understood through Peter Thiel’s concept of definite and indefinite optimism. In Thiel’s dichotomy, indefinite optimists believe the world will get better but have no idea how, whereas definite optimists have a concrete proposal for how to make the world better.
How does this connect to finite and infinite games? Paradoxically, indefinite optimism leads to an obsession with finite games, since there’s no higher animating principle at work to drive progress. When you don’t have any positive vision for innovation, the natural solution is to write procedures and hope that progress will arise spontaneously if the right steps are followed. Companies, research groups, and other organizations can learn to mimic what actual innovation looks like from afar, but without the proper motivations their ultimate success is improbable.
In contrast, an organization playing an infinite game doesn’t need to be forced to jump through arbitrary hoops. Pharmaceutical companies don’t bother to acquire 13C NMR and IR spectra for every intermediate; startups putting together a minimum viable product don’t worry about properly formatting all their code. Finite games are only a distraction for properly motivated organizations, and one which should be avoided whenever possible.1
What conclusions can we draw from this? On a personal level, seek to make your games as infinite as possible. Every startup has to raise money and every graduate student has to publish papers, but one shouldn’t spend most of one’s time worrying about how to publish papers as efficiently as possible.
If you can treat finite games as the distraction that they are, you can give them as little mental effort as possible and spend your time and talents on worthier pursuits.
On a broader level, I’m struck by the fact that finite games are a problem of our own making. Nobody becomes a scientist hoping to write papers or win grants;2
our aspirations start out infinite, and it’s only through exposure to the paradigms of the field that we learn to decrease our ambition.
Indeed, calling a research proposal “ambitious” is reportedly one of the worst criticisms that an NIH study section can give.
We should therefore be skeptical about bureaucratic solutions to research stagnation. If our scientific institutions themselves are part of the problem, expanding their reach and importance is unlikely to fix the problem and may indeed do more harm than good. “If you find yourself in a hole, stop digging.”
Thanks to Jacob Thackston and Ari Wagen for reading drafts of this piece.
Footnotes
I’m intentionally eliding the normative question of “how do we assign funding if not through quantitative measures?”—it’s an excellent question, and one which deserves a larger treatment than I can offer here. The Nielsen/Qiu metascience essay has some ideas here.
This may not always be true, but I think it’s generally true. Most high schoolers or undergraduates are drawn to science because of a sense of wonder or curiosity, which gets transmuted to “publish JACS papers” through the alchemy of graduate school.
Ari pointed me towards this essay by a physics graduate student, which seems relevant.
Modeling ion-pair association/dissociation is an incredibly complex problem, and one that's often beyond the scope of conventional DFT-based techniques.
(This study is a nice demonstration of how hard modeling ion-pairing can be.)
Nevertheless, it's still often important to gain insight into what the relative energy of solvent-separated ion pairs and contact-ion pairs might be;
are solvent-separated configurations energetically accessible or not?
I've run into this problem a few times myself: in measuring pKas in nonpolar solvents, and again recently when trying to understand
the solution structure of ethereal HCl.
When digging through the pKa literature, I was surprised to learn that there's a simple and relatively accurate way to estimate the dissociation constant of contact-ion pairs,
developed by Raymond Fuoss in the 1950s.
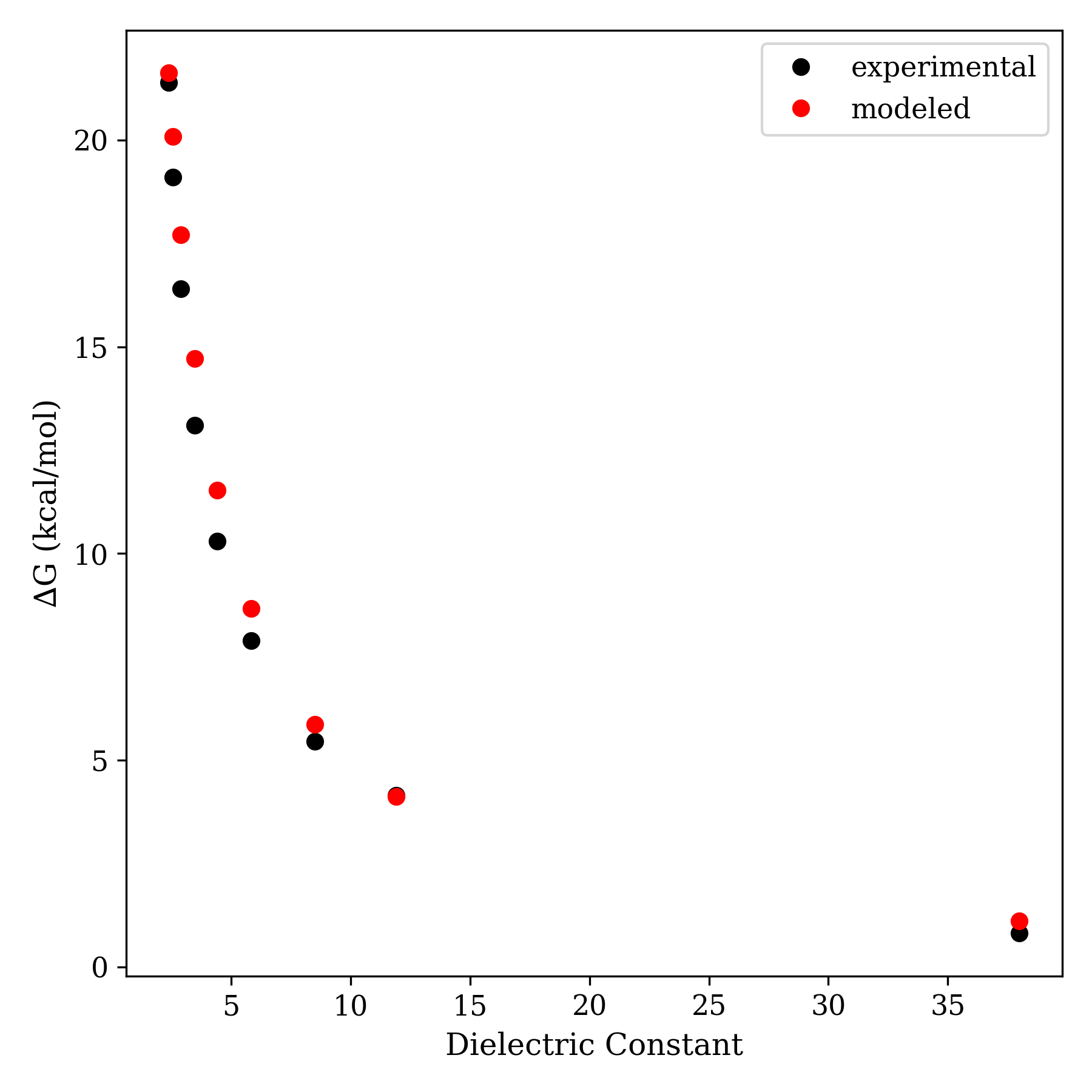
Despite modeling ions as charged spheres and solvent as a featureless dielectric, the "Fuoss model" is surprisingly good at reproducing experimental data.
Here's one such example:
Comparison of experimental ∆Gdiss for tetraisoamylammonium nitrate in various dioxane–water blends vs. Fuoss-calculated ∆G values.
(Data taken from Fuoss, J. Am. Chem. Soc., 1933, Table II.)
It's not trivial to think about how to get similar results using more atomistic methods!
(In principle one could actually model the exact solvent mixture and compute the energy of ion-pair dissociation using biased sampling and MD, but this
would be horrendously expensive and probably less accurate anyway.)
The Fuoss model is pretty simple to implement oneself—but to make things even easier, I've implemented it as an open-source Python package,
which can be imported using pip.
The package contains only a single function, compute_kd, which accepts the name of the cation, the name of the anion, and the dielectric constant of the medium.
(Alternatively, .xyz files, .gjf files, or cctk.Molecule objects can also be given.)
Under the hood, the program builds molecules using cctk, computes their volume, and then applies the Fuoss model.
The end result is a comically simple interface:
On the associated Github repository, there's also a little command-line script which makes this even simpler:
$ python quick_fuoss.py tetraisoamylammonium nitrate 8.5
Reading ion #1 from rdkit...
Reading ion #2 from rdkit...
Dissociation constant: 0.00004930 M
Ionization energy: 5.873 kcal/mol
$ python quick_fuoss.py tetraisoamylammonium nitrate 11.9
Reading ion #1 from rdkit...
Reading ion #2 from rdkit...
Dissociation constant: 0.00094706 M
Ionization energy: 4.122 kcal/mol
My hope is that this program promotes wider adoption of the Fuoss model, and in general enables more critical thinking about ion-pair energetics in organic solvents.
Please feel free to send any bug reports, complaints, etc. my way!
Last week, I posted a simple Lennard–Jones simulation, written in C++, that models the behavior of liquid Ar in only 1561 characters.
By popular request, I'm now posting a breakdown/explanation of this program, both to explain the underlying algorithm and illustrate how it can be compressed.
Here's the most current version of the program, now condensed even further to 1295 characters:
Let's add some whitespace and break this down, line by line:
#include <iostream>
#include <random>
// some abbreviations
#define H return
typedef double d;
typedef int i;
using namespace std;
// constants
i N=860; // number of particles
d B=8.314e-7; // boltzmann's constant
d S=1544.8; // the minimum of the Lennard-Jones potential, 3.4 Å, to the 6th power
The program begins by importing the necessary packages, defining abbreviations, and declaring some constants.
Redefining double and int saves a ton of space, as we'll see.
(This is standard practice in the code abbreviation world.)
// given a 1d coordinate, scale it to within [-17,17].
// (this only works for numbers within [-51,51] but that's fine for this application)
d q(d x){
if(x>17)
x-=34;
if(x<-17)
x+=34;
H x;
}
// returns a uniform random number in [-17,17]
d rd(){
H 34*drand48()-17;
}
We now define a helper function to keep coordinates within the boundaries of our cubical box, and create another function to "randomly" initialize particles' positions.
(drand48 is not a particularly good random-number generator, but it has a short name and works well enough.)
// vector class
struct v{
d x,y,z;
v(d a=0,d b=0,d c=0):x(a),y(b),z(c){}
// return the squared length of the vector
d n(){
H x*x+y*y+z*z;
}
// return a vector with periodic boundary conditions applied
v p(){
H v(q(x),q(y),q(z));
}
};
// vector addition
v operator+(v a,v b){
H v(a.x+b.x,a.y+b.y,a.z+b.z);
}
// multiplication by a scalar
v operator*(d a,v b){
H v(a*b.x,a*b.y,a*b.z);
}
Here, we define a vector class to store positions, velocities, and accelerations, and define addition and multiplication by a scalar.
(It would be better to pass const references to the operators, but it takes too many characters.)
// particle class
struct p{
// r is position, q is velocity, a is acceleration, b is acceleration in the next timestep
v r,q,a,b;
// neighbor list, the list of particles close enough to consider computing interactions with
vector<i> W;
p(v l):r(l){}
// apply a force to this particle.
// 0.025 = 1/40 = 1/(Ar mass)
void F(v f){
b=0.025*f+b;
}
};
// global vector of all particles
vector<p> P;
// write current coordinates to stdout
void w(){
cout<<N<<"\n\n";
for(p l:P)
cout<<"Ar "<<l.r.x<<" "<<l.r.y<<" "<<l.r.z<<"\n";
cout<<"\n";
}
Now, we define a class that represents a single argon atom.
Each atom has an associated position, velocity, and acceleration, as well as b, which accumulates acceleration for the next timestep.
Atoms also have a "neighbor list", or a list of all the particles close enough to be considered in force calculations.
(To prevent double counting, each neighbor list only contains the particles with index larger than the current particle's index.)
We create a global variable to store all of the particles, and create a function to report the current state of this variable.
// compute forces between all particles
void E(){
for(i j=N;j--;){
for(i k:P[j].W){
// compute distance between particles
v R=(P[j].r+-1*P[k].r).p();
d r=R.n();
// if squared distance less than 70 (approximately 6 * 3.4Å**2), the interaction will be non-negligible
if(r<70){
d O=r*r*r;
// this is the expression for the lennard–jones force.
// the second lennard–jones parameter, the depth of the potential well (120 kB), is factored in here.
R=2880*B*(2*S*S/O/O-S/O)/r*R;
// apply force to each particle
P[j].F(R);
P[k].F(-1*R);
}
}
}
}
Now, we create a function to calculate the forces between all pairs of particles.
For each particle, we loop over the neighbor list and see if the distance is within six minima of the adjacent particle,
using squared distance to avoid the expensive square-root calculation.
If so, we calculate the force and apply it to each particle.
// build neighbor lists
void e(){
for(i j=0;j<N;j++){
// clear the old lists
P[j].W.clear();
for(i k=j+1;k<N;k++)
// if squared distance between particles less than 90 (e.g. close to above cutoff), add to neighbor list
if((P[j].r+-1*P[k].r).p().n()<90)
P[j].W.push_back(k);
}
}
Finally, we create a function to build the neighbor lists, which is a straightforward double loop.
We add every particle which might conceivably be close enough to factor into the force calculations within 10–20 frames.
i main(){
i A=1e3; // initial temperature (1000 K)
i Y=5e3; // time to reach final temperature (5000 fs)
// initialize the system. each particle will be randomly placed until it isn't too close to other particles.
for(i j=N;j--;){
for(i a=999;a--;){
// generate random position
v r=v(rd(),rd(),rd());
i c=0;
// check for clashes with each extant particle
for(p X:P){
d D=(r+-1*X.r).p().n();
if(D<6.8)
c=1;
}
// if no clashes, add particle to list
if(!c){
P.push_back(p(r));
break;
}
}
}
To begin the program, we randomly initialize each particle and test if we're too close to any other particles.
If not, we save the position and move on to the next particle. This is crude but works well enough for such a simple system.
// run MD! this is basically just the velocity verlet algorithm.
for(i t=0;t<=3e4;t++){
// update position
for(p& I:P)
I.r=(I.r+I.q+0.5*I.a).p();
// every 20 timesteps (20 fs), update neighbor lists
if(t%20==0)
e();
// compute forces
E();
// finish velocity verlet, and sum up the kinetic energy.
d K=0; // kinetic energy
for(p& I:P){
I.q=I.q+0.5*(I.b+I.a);
I.a=I.b;
I.b=v();
K+=20*I.q.n();
}
d T=2*K/(3*B*N); // temperature
// in the first 10 ps, apply berendsen thermostat to control temperature
if(t<2*Y){
d C=75; // target temperature
if(t<Y)
C=75*t/Y+(A-75)*(Y-t)/Y;
for(p& I:P)
I.q=sqrt(C/T)*I.q;
}
// every 100 fs after the first 10 ps, write the configuration to stdout
if(t%100==0&&t>2*Y)
w();
}
}
Finally, we simply have to run the simulation. We use the velocity Verlet algorithm with a 1 fs timestep,
updating neighbor lists and writing to stdout periodically.
The temperature is gradually lowered from 1000 K to 75 K over 5 ps, and temperature is controlled for the first 10 ps.
Hopefully this helps to shed some light on how simple a molecular dynamics simulation can be, and highlights the wonders of obfuscated C++!