I first encountered organic chemistry on Wikipedia, my freshman year of high school. The complexity and arcanity of the field instantly attracted me: here was something interesting that I didn’t know about and which didn’t require years of mathematical training to approach (unlike most of physics).
I soon started reading about organic chemistry more and more, albeit with no rhyme or reason to my study. I didn’t know what the good textbooks were, what order to study things in or which concepts ought to be understood in depth before progressing further. Organic chemistry was just a “glass bead game” to me, an art of symbols devoid of any real-world representations. But enthusiasm can sometimes suffice where wisdom is lacking.
With the support of a teacher and a half-dozen friends who also wanted to learn more organic chemistry, we started a little independent study. We met in a closet and read a textbook, worked through the problems, and our teacher wrote us tests. We eventually managed to get through all of Paula Bruice, although various misconceptions (and mispronunciations) stayed with me until I took Movassaghi’s course at MIT.
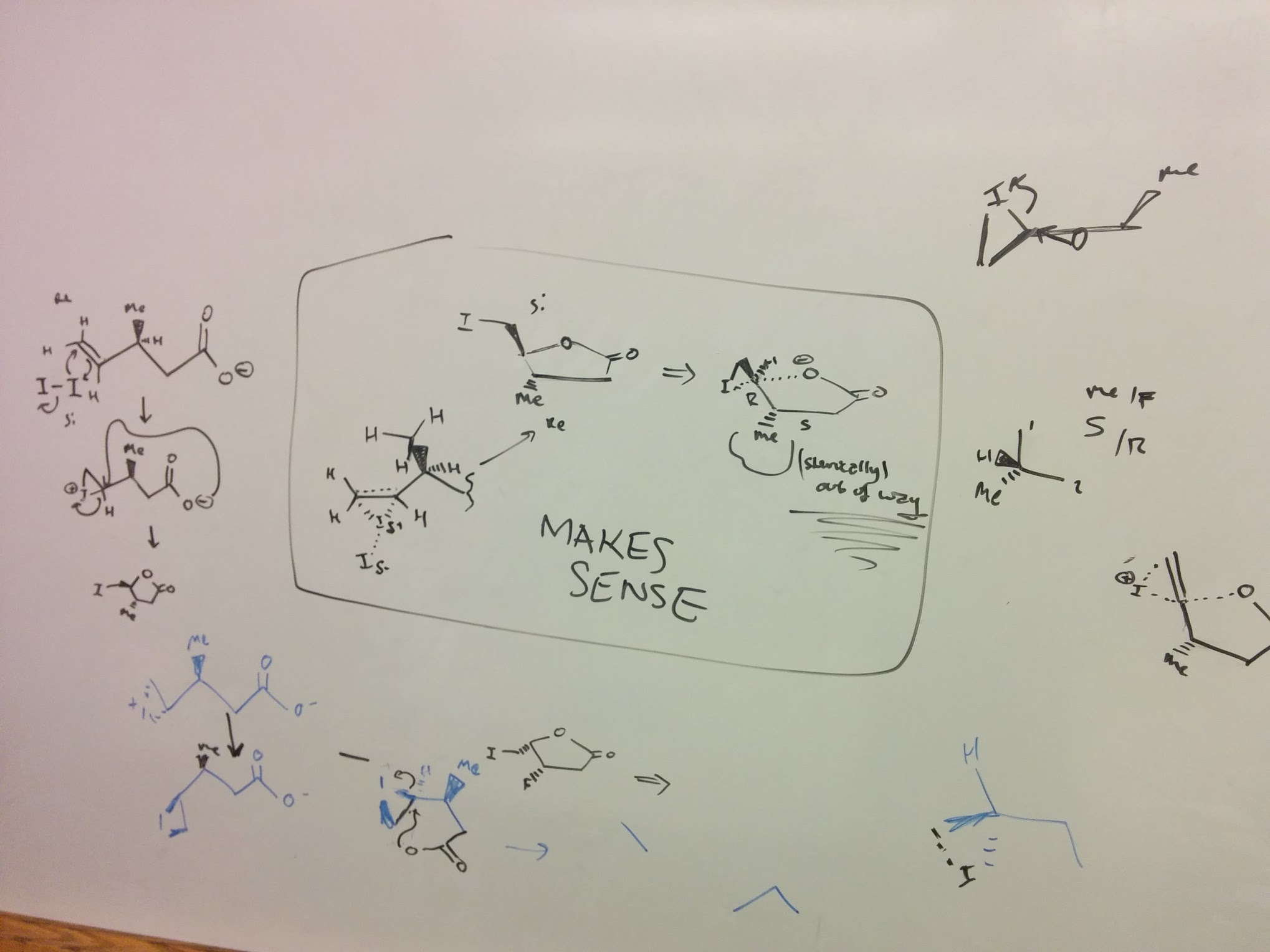
Here’s a picture of some work from high school, which does not in fact make very much sense. (I think this is from the Evans problem set library.)
But in high school we had no lab, and thus no practical knowledge. We split into two groups and tried to come up with experiments for ourselves, but the results were dismal. Here’s the procedure (copied verbatim) for the one and only reaction we ran, synthesis of nitrobenzene via nitration of benzene, which was to be the first step in a multistep synthesis of Kevlar:
In a 250 mL beaker, dissolve benzene in a solution of concentrated H2SO4 of twice its volume. Use an ice-salt bath to bring this solution to 0 °C or below, and use a Pasteur pipette to slowly add a 1:1 solution of HNO3 and H2SO4 (be sure to keep the solution below 15 degrees Celsius at all times, as the reaction is strongly exothermic). Once all the solution has been added, warm it to room temperature and allow it to sit for 15 minutes.
Pour the solution over 50 g of crushed ice in a 250 mL beaker. Once the ice has melted, isolate the product via vacuum filtration via a Buchner funnel and rinse it twice with water and twice with methanol. Recrystallize in a solution of methanol.
The astute observer will notice that there aren’t very many details here. How much benzene? How much nitric acid? We didn’t have the glassware mentioned above—neither a beaker nor a Buchner funnel. And, perhaps most damningly, the procedure calls for isolation by filtration, challenging since nitrobenzene is a liquid at room temperature.
Despite these problems, we successfully ran this reaction (open to air, not in a fume hood), and obtained the product. I vividly recall the yellow bubbles of nitrobenzene floating to the top of the vial, and the smell of cherries that filled the room, a smell that returns to me in Proustian fashion from time to time when using certain reagents. We didn’t have a separatory funnel (or we didn’t know how to use it if we did), so we fished some of the nitrobenzene out with a Pasteur pipette and threw the rest away.
A little bit of knowledge would have served us well, as would have gloves and a fume hood. Here’s Wikipedia:
Prolonged exposure [to nitrobenzene] may cause serious damage to the central nervous system, impair vision, cause liver or kidney damage, anemia and lung irritation. Inhalation of vapors may induce headache, nausea, fatigue, dizziness, cyanosis, weakness in the arms and legs, and in rare cases may be fatal…
Nitrobenzene is considered a likely human carcinogen by the United States Environmental Protection Agency, and is classified by the IARC as a Group 2B carcinogen which is "possibly carcinogenic to humans".
Indeed, a coworker of mine would later be sent to the hospital after spilling nitrobenzene on herself. Surprisingly, my group’s experiment still ended up being the safer one—the lab portion of our course was disbanded the following day after my classmates caused an explosion with thionyl chloride.
I was fortunate enough to land a summer research internship in the Sessler lab at UT the following summer, and I started studying chemistry in earnest: column chromatography, Anslyn/Dougherty, NMR spectroscopy, and all the rest. I remember the first reaction I ran at UT: retro-Friedel–Crafts dealkylation of a calix[4]arene, using about 10 g each of phenol and aluminum(III) chloride. The brutal physicality of lab work was a nice contrast to the gnosticism of software (where I’d worked previously), and I was hooked.
I’ll fast-forward through the more recent parts of my chemical career: I went to MIT and joined the Imperiali lab to work on essentially a medicinal chemistry project: hit-to-lead optimization, featuring lots of de novo heterocycle synthesis. I got to cook reactions in molten urea, quench 500 mL of phosphorus oxychloride at a time, and even design new routes (with a little oversight from my postdoc). It was awesome.
My hood in the Imperiali Group. Although this hood appears to have a Schlenk line, do not be fooled—it’s purely decorative.
After three semesters, I got tired of my cross couplings mysteriously failing and joined the Buchwald lab, where I learned how to do chemistry more carefully: handling air-sensitive materials with a Schlenk line or in the glovebox, not “as fast as possible.” My tenure in the Buchwald lab also introduced me to the importance of computations, which became a key part of my doctoral work, particularly when we were sent home in March of my first year. COVID gave me the opportunity to pursue some software engineering projects that I wouldn’t have had time to work on otherwise (like cctk and presto), and simulation started to take up more and more of my time and intellectual bandwidth.
A large-scale biaryl synthesis proceeding through a benzyne intermediate, from my time in the Buchwald Group.
One of my first Gaussian jobs ever. I didn't get cluster access for a little while, so I used to run Gaussian on the printer computers in my dorm's basement and just hope that nobody needed the computer overnight.
I defended my dissertation on June 5th, and cleaned out my hood last week. For the foreseeable future, I’ll be a purely computational chemist—computation is advancing quickly, and I think that’s where I have the most to offer the field right now. But I’ll miss the sights and sounds of the lab. There’s a satisfaction to making a new molecule and holding the final product in your hands: the knowledge that you’ve reshaped this little corner of reality through your own actions, and that this particular arrangement of matter has never existed before.
And simulation is, at the end of the day, only useful insofar as it helps us make real molecules. There may be people who wish to model reactions purely for the sake of modeling them, but I am not one of them. What drew me to simulation initially, and what still attracts me to the field today, is its potential to help experimental scientists do their work faster and better. It was easy for me to ensure that this was true when I was both doing the experiments and running the simulations; my incentives were aligned properly. It will be harder in the future.
There’s a seductive appeal in leaving lab work behind altogether, too, and one that’s dangerous. Any experimentalist who’s worked with a computational collaborator knows that nothing ever works precisely as modeled. There are untold depths of chemical behavior still inaccessible to the idealized world of simulation, and it’s all too easy for computational chemists just to look the other way. Life is easier when you don’t have to deal with sludgy workups or poorly soluble intermediates, but they don’t go away just because you can’t model them by DFT—reality has a way of keeping us honest that simulations frequently lack.
So, although I bid lab farewell at this point in my career, it’s a bittersweet parting. I hope to return someday; only time will tell.
Thanks to Jacob Thackston for reading a draft of this piece.
I started this blog one year ago today, with a post on site-selective glycosylation. According to Google Analytics, there have been 24,035 views since then.
The only of these that really surprises me is #5: the 13C NMR post made a lot of organic chemists really angry, the Talent review was reposted on Marginal Revolution, and Delian Asparouhov (CEO of Varda) retweeted my post about Varda’s crystallization ideas. And everyone loves to share a ranking of the year’s papers, especially when their own work is highlighted.
Twitter downranked the Substack post pretty heavily, so it’s not surprising that nobody saw it. The Lennard–Jones posts are more unexpected. Whenever I write about anything computational or coding-related, it seems to attract much less engagement, which is perhaps a reflection of the fact that most of my followers are experimental chemists who don’t really care about obfuscated C code.
A year in, writing blog posts has gotten much easier. The following advice from Alexey Guzey didn’t seem true when I started, but it does seem true now:
Writing not only helps you to understand what’s going on and to crystallize your thoughts, it actually makes you think of new ideas and come up with solutions to your problems.
I’ve fallen into a 1x/week update schedule, which seems to work pretty well: enough to keep the routine up, but not so much that it’s a serious distraction from my actual job. I hope to maintain this schedule for the foreseeable future, and recommend it to other bivocational bloggers.
Anyhow, thanks for reading!
(Also, today in off-blog content: I appeared on my first podcast, Forbidden Conversations, hosted by Harry Wetherall. We talk about why people don’t have kids earlier, how I reconcile being a Christian with being a scientist, the concept of “cope,” and more: you can check it out on Apple Podcasts or Spotify.)
Not Boring recently published a panegyric about Varda, a startup that’s trying to create “space factories making drugs in orbit.” When I first read this description, alarm bells went off in my head—why would anyone try to make drugs in space? Nevertheless, there’s more to this idea than I initially thought. In this piece, I want to dig a little deeper into the chemistry behind Varda, and discuss some potential advantages and challenges of the approach they’re exploring.
Much of my confusion was quickly resolved by realizing that Varda is not actually “making drugs in orbit,” or not in the way that an organic chemist would interpret that sentence. Varda’s proposal is actually much more specific: they aim to crystallize active pharmaceutical ingredients (APIs, i.e. finished drug molecules) in microgravity, allowing them to access crystal forms and particle size distributions which can’t be made under terrestrial conditions. To quote from their website:
Varda’s microgravity platform is grounded in decades of proven research conducted on NASA’s space stations. By eliminating factors such as natural convection and sedimentation, processing in a microgravity environment provides a unique path to formulating small molecules and biologics that traditional manufacturing processes cannot address. The resulting tunable particle size distributions, more ordered crystals, and new forms can lead to improved bioavailability, extended shelf-life, new intellectual property, and new routes of administration.
Crystallization is an excellent target for a new and expensive manufacturing process because it’s at once very important and very hard to control. The goal of crystallization is to grow crystals of a given compound, fitting the component molecules together into a perfect lattice that excludes impurities and can be easily handled. To the best of my knowledge, almost every small-molecule API is crystallized at one stage or another; it’s the best way to ensure that the material is extremely pure.
(Crystallizing proteins for administration is less common, since proteins are really difficult to crystallize, but it’s not unheard of—insulin is often administered subcutaneously as a microcrystalline suspension, which allows higher concentrations to be accessed without excessive viscosity.)
But crystallization is also something we can’t really control. We can’t physically put molecules into a lattice or force them to adopt ordered configurations; all we can do is dissolve them in some mixture, tweak the conditions a little bit, and hope that crystals form. Thus, finding good crystallization conditions basically amounts to randomly screening solvents and additives, leaving the solutions for a long time, and checking to see if crystals grow. In the words of Derek Lowe:
I'd like to open up the floor for nominations for the Blackest Art in All of Chemistry. And my candidate is a strong, strong contender: crystallization. When you go into a protein crystallography lab and see stack after stack after stack of plastic trays, each containing scores of different little wells, each with a slight variation on the conditions, you realize that you're looking at something that we just don't understand very well.
Why does microgravity matter for crystallization? Not Boring says that crystallization occurs “at the mesoscopic scale, the length scale on which objects are larger than nanoscale (on the order of atoms and molecules) but still small enough that quantum mechanical or other non-trivial ‘microscopic’ behavior becomes apparent.” I found this answer a little confusing—doesn’t crystallization begin on the nanoscale and end on the macroscopic scale?
Clearer to me was the explanation from a 2001 review by Kundrot and co-workers:
In zero gravity, a crystal is subject to Brownian motion as on the ground, but unlike the ground case, there is no acceleration inducing it to sediment [fall out of solution]. A growing crystal in zero gravity will move very little with respect to the surrounding fluid. Moreover, as growth units leave solution and are added to the crystal, a region of solution depleted in protein is formed. Usually this solution has a lower density than the bulk solution and will convect upward in a 1g field as seen by Schlerien photography (Figure 1). In zero gravity, the bouyant [sic] force is eliminated and no bouyancy-driven convection occurs. Because the positions of the crystal and its depletion zone are stable, the crystal can grow under conditions where its growing surface is in contact with a solution that is only slightly supersaturated. In contrast, the sedimentation and convection that occur under 1g place the growing crystal surface in contact with bulk solution that is typically several times supersaturated. Lower supersaturation at the growing crystal surface allows more high-energy misincorporated growth units to disassociate from the crystal before becoming trapped in the crystal by the addition of other growth units…
In short, promotion of a stable depletion zone in microgravity is postulated to provide a better ordered crystal lattice and benefit the crystal growth process.
To summarize, microgravity serves to immobilize the crystal with respect to the surrounding solution, preventing convection or sedimentation from bringing highly concentrated solutions into contact with the crystal. This slows crystal growth, which might sound bad but is actually really good: in general, the slower a crystal grows, the higher its purity. (See also this 2015 article for further discussion.)
What practical impact does this have? In most cases, crystals grown in space are better than their terrestrial congeners by a variety of metrics: larger, structurally better, and more uniform. To quote from the Not Boring piece:
Doing crystallization in space is like adding a gravity knob to your instrument—it opens up regions of process design space that would otherwise be inaccessible. Importantly, after the crystallization occurs in space, the drug retains its solid state upon re-entry. Manufacture in space; use on earth.
This is why pharma is going to space to experiment with a wide range of medicines. Formulations made in microgravity could open the door to improvements in drug shelf life, bioavailability, IP expansion, and even better approaches to drug delivery…
To date, there has been a major disconnect between microgravity research and manufacturing. While it’s been possible to hitch a ride to the ISS and collaborate with NASA on PCG experiments, there is no existing commercial offering to actually manufacture drugs in space. Merck used their research results on Keytruda® crystallization to tinker with their terrestrial approaches to formulation. What if they could actually just manufacture the crystals they discovered in microgravity at commercial scale?
This is Varda’s mission—to make widespread research and manufacturing in microgravity a reality.
One concern I have is that to date, the vast majority of space-based crystallization has been aimed at structural biology (elucidating the structure of a protein via crystallography), which only takes one crystal, one time. What Varda is aiming to do is preparative crystallography: crystallizing proteins and small molecules to isolate large quantities of them. Both processes obviously involve growing crystals, but otherwise they’re pretty different: in structural biology, all you care about is isolating a single large and very pure crystal, while uniformity and reproducibility are paramount in preparative crystallography.
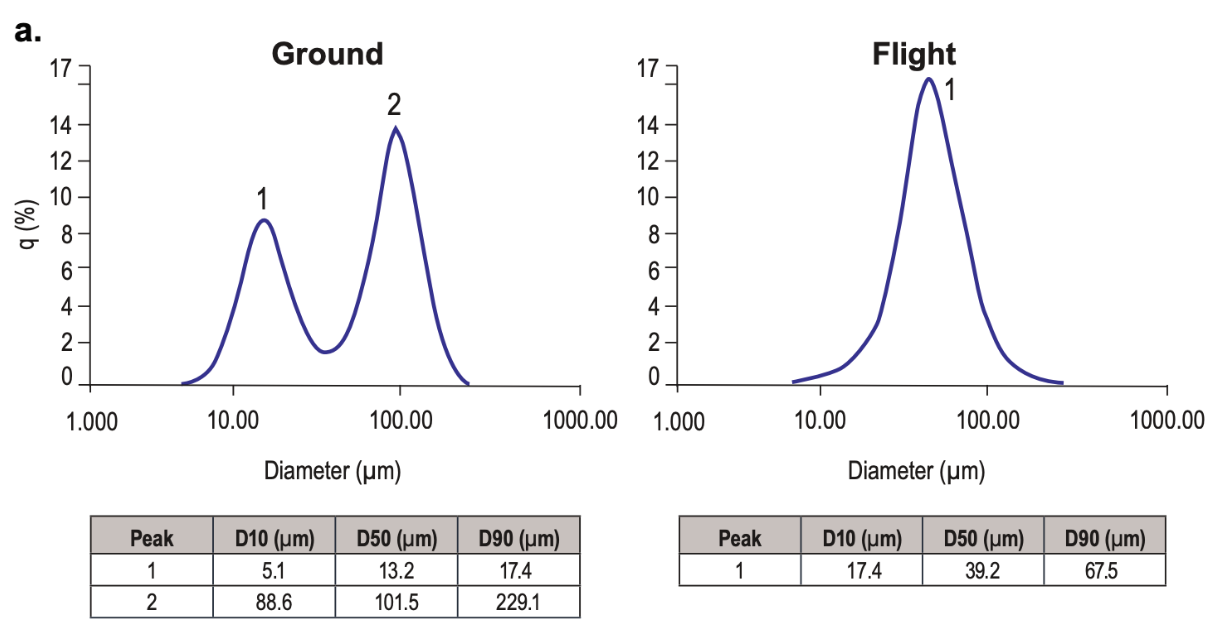
There’s some precedent for preparative protein crystallization in microgravity: a 1996 Schering-Plough paper studied crystallization of zinc interferon-α2b on the Space Shuttle. The results are excellent: over 95% of the protein crystallized, and the resulting suspension showed good stability and improved serum half-life in Cynomolgus monkeys (ref, ref). The difference in crystal quality is huge:
Space-grown crystals are much, much larger than crystals grown on Earth.
More recently, scientists from Merck found that crystallization of pembrolizumab (Keytruda) in microgravity reproducibly formed a monomodal particle size distribution, as opposed to the bimodal particle size distribution formed under conventional conditions (ref), although the crystals didn’t seem any larger:
Comparison of pembrolizumab crystal size distribution under different conditions.
Of course, until recently access to space was very limited and there wasn’t much reason to study preparative protein crystallography in microgravity, so the lack of studies is hardly surprising. For all the reasons discussed above, it seems very likely that preparative crystallography will be generally better in microgravity, and that the resulting crystals will be more homogenous and more pure than the crystals you could grow on Earth. But it’s not 100% certain, and that’s something Varda will have to establish. The word “can” is doing a lot of work in this text from their website:
By eliminating factors such as natural convection and sedimentation, processing in a microgravity environment provides a unique path to formulating small molecules and biologics that traditional manufacturing processes cannot address. The resulting tunable particle size distributions, more ordered crystals, and new forms can lead to improved bioavailability, extended shelf-life, new intellectual property, and new routes of administration (emphasis added)
Another concern is that little work (that I’m aware of) has been done on small molecule crystallization in microgravity—the very task Varda intends to start with. Small molecules, in general, are much easier to crystallize than proteins, and there are more parameters for the experimental scientist to tune. While there are certainly cases where small molecule crystals can display unexpected or problematic behaviors (like the famous case of ritonavir, the molecule Varda is investigating first), in general crystallization of small molecules seems like an easier problem, and one for which there are better state-of-the-art workarounds.
The most likely failure mode to me, though, is just that microgravity crystallization is better than crystallization on Earth, but not that much better. Shooting APIs into space, waiting for them to crystallize, and then launching them back to Earth is going to be really expensive, and Varda will have to demonstrate extraordinary results to justify the added hassle—particularly for a technique that they hope to make a key part of the pharmaceutical manufacturing process. Talk about supply chain risk! (Is this all going to be GMP?)
And it’s worth pointing out a fairly obvious consideration too: what Varda is proposing to do in space is only one part of a vast, multi-step operation. No synthesis will take place in space, so all the manufacturing of either proteins or small molecules will take place on Earth. The purified products will then be launched into space, crystallized, and then the crystal-containing solution will undergo reentry and then be formulated into whatever final form the drug needs to be administered in.
So, although “there are a number of small molecules that fetch hundreds of thousands or millions of dollars per kg” (Not Boring), Varda can address only a small—albeit important—part of the manufacturing process. I doubt this is likely to change. On average, it takes 100–1000 kg of raw materials to manufacture 1 kg of a small molecule drug (ref), so shipping everything to orbit would massively raise costs, without any obvious advantages that I can think of. The TAM for Varda might be large enough to break even, but it’s not going to replace conventional pharmaceutical manufacturing anytime soon.
Varda’s pitch is perfect for venture capital: ambitious, risky, and potentially game-changing if it succeeds. And I wish them luck in their quest, since new and better ways to approach formulation would be awesome. But I can’t shake the nagging doubt that they’re so excited about the image of space-based manufacturing that they’re trying to invent a problem that their aerospace engineers have a solution for. We’ll find out soon enough if they’ve succeeded.
Screening on only one substrate before assessing the substrate scope. This is the “ordinary means” in methods development.
Screening on one substrate, but choosing a substrate that worked poorly in a previous study (e.g.). This can be thought of as serial multi-substrate screening, where each substrate is a separate project, but the body of work achieves greater generality over time.
Screening on one substrate at a time, but rescreening catalysts when you find problematic substrates (e.g.). This amounts to serial multi-substrate screening within a single project.
Intentionally choosing a variety of catalysts up front and screening this set of catalysts for each new substrate class (e.g.), thus achieving a high degree of generality with a family of catalysts, but without attempting to systematically quantify the generality of each catalyst in this set.
Choosing a handful of model substrates instead of just one, but otherwise doing everything the same as one would normally (e.g., pages S24–S29).
Intentionally choosing a large, diverse panel of substrates and screening against this panel to quantify catalyst generality over a given region of chemical space. This is essentially what we and the Miller group did recently (and others, etc).
The same, but incorporating robotics, fancy analytical methods, generative AI, or whatever else.
When I present the “screening for generality” work, I often get the response “this is cool, but my reaction doesn’t work in 96-well plates/I don’t have an SFC-MS/my substrates are hard to make.” The point of this taxonomy is to illustrate that there are a lot of ways to move towards “screening for generality” that don’t involve 96-well plates.
If you have the time and resources for robotics or SFC-MS, that’s great—you’ll be able to screen more quickly and cover more ground. But you can still start to consider more than a single model substrate even without any specialized equipment. It’s a mindset, not a recipe.
Much ink has been spilled on whether scientific progress is slowing down or not (e.g.). I don’t want to wade into that debate today—instead, I want to argue that, regardless of the rate of new discoveries, acquiring scientific data is easier now than it ever has been.
There are a lot of ways one could try to defend this point; I’ve chosen some representative anecdotes from my own field (organic chemistry), but I’m sure scientists in other fields could find examples closer to home.
NMR Spectroscopy
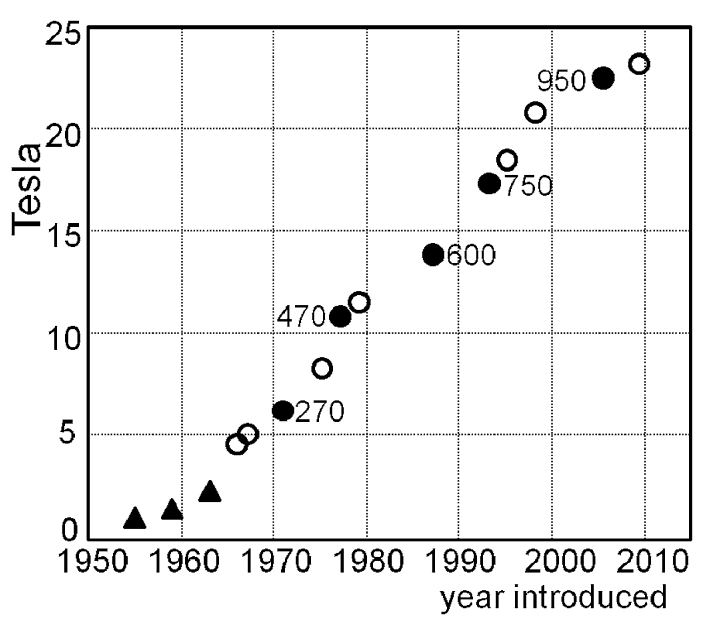
NMR spectroscopy is now the primary method for characterization and structural study of organic molecules, but it wasn’t always very good. The last half-century has seen a steady increase in the quality of NMR spectrometers (principally driven by the development of more powerful magnetic fields), meaning that even a relatively lackluster NMR facility today has equipment beyond the wildest dreams of a scientist in the 1980s:
Most powerful NMR magnet over time (Campbell, Figure 1). Broad adoption lags these records, but the trend is comparable.
Commercially Available Compounds
The number of compounds available for purchase has markedly increased in recent years. In the 1950s, the Aldrich Chemical Company’s listings could fit on a single page, and even by the 1970s Sigma-Aldrich only sold 40,000 or so chemicals (source).
But things have changed. Nowadays new reagents are available for purchase within weeks or months of being reported (e.g.), and companies like Enamine spend all their time devising new and quirky structures for medicinal chemists to buy.
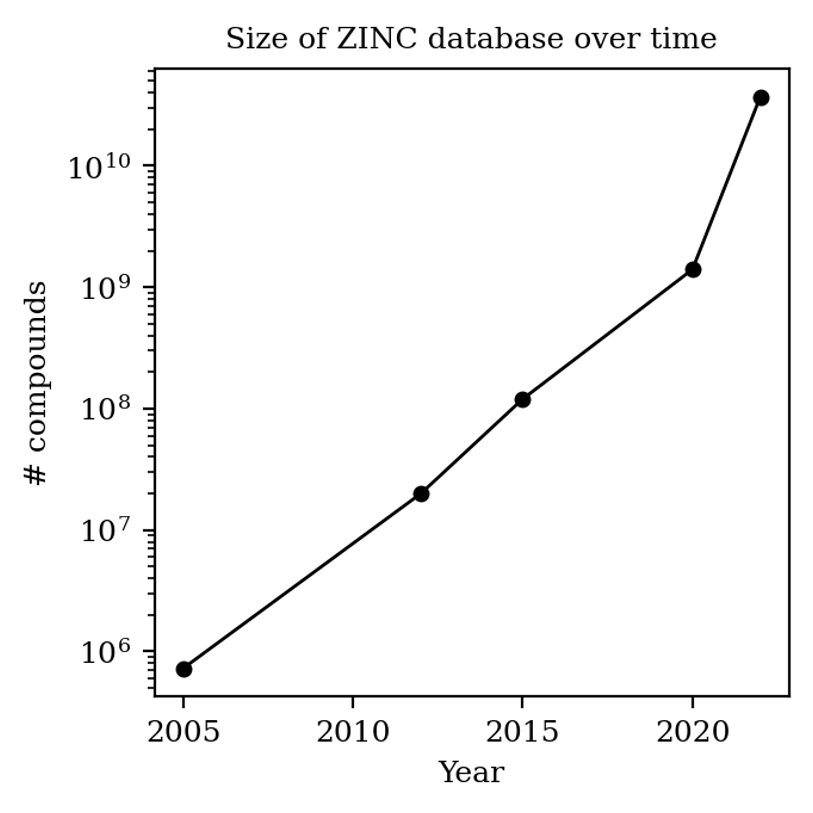
One way to quantify how many compounds are for sale today is through the ZINC database, aimed at collecting compounds for virtual screening, which is updated every few years or so. The first iteration of ZINC, in 2005, had fewer than a million compounds: now there are almost 40 billion:
Number of compounds in the ZINC database over time (graphic made by me).
(Most compounds in the ZINC database aren’t available on synthesis scale, so it’s not like you can order a gram of all 40 billion compounds—there’s probably more like 3 million “building blocks” today, which is still a lot more than 40,000.)
Chromatography
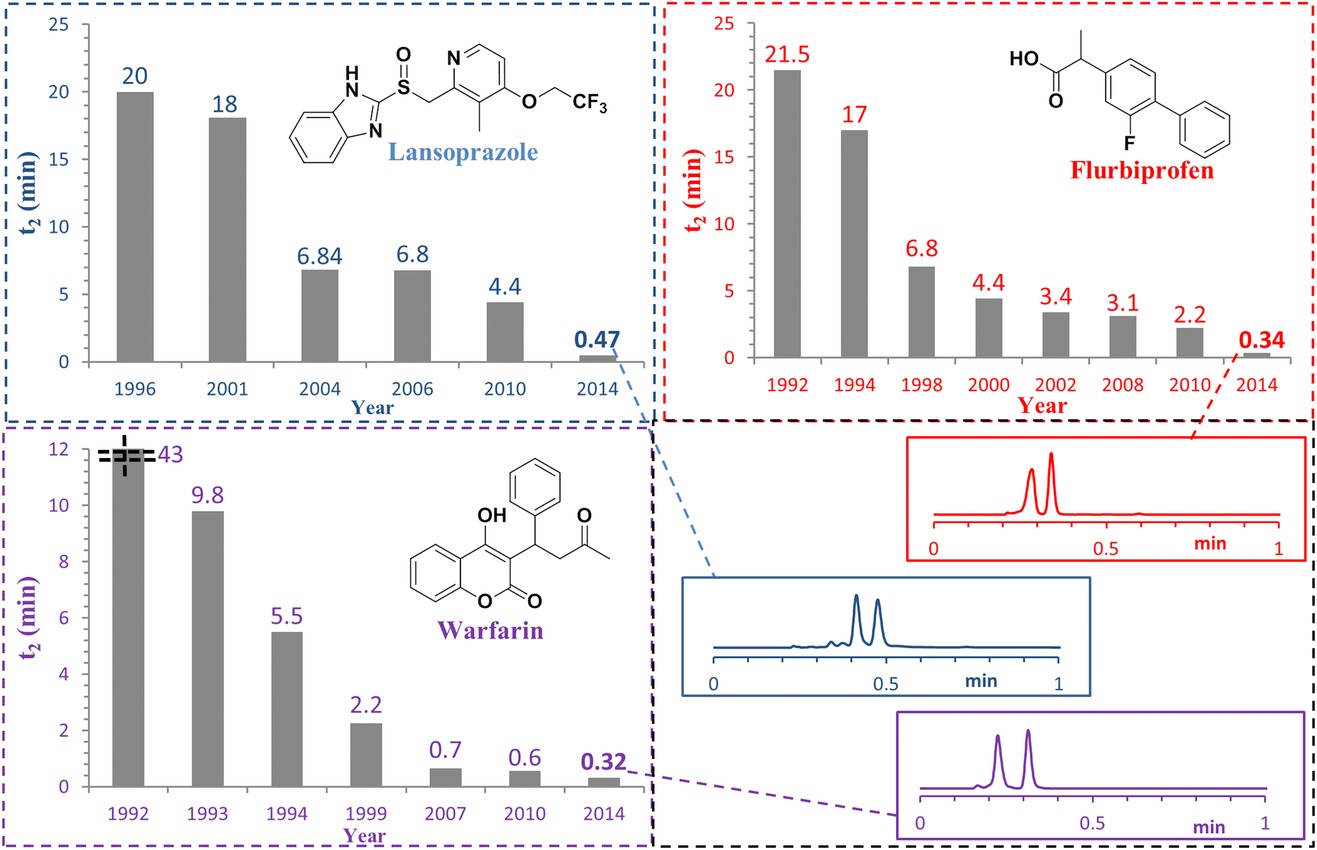
Chromatography, the workhorse of synthetic and analytical chemistry, has also gotten a lot better. Separations that took almost an hour can now be performed in well under a minute, accelerating purification and analysis of any new material (this paper focuses on chiral stationary phase chromatography, but many of the advances translate to other forms of chromatography too).
Fastest chiral separation for a given compound by year (Regalado, Figure 1).
Computational Chemistry
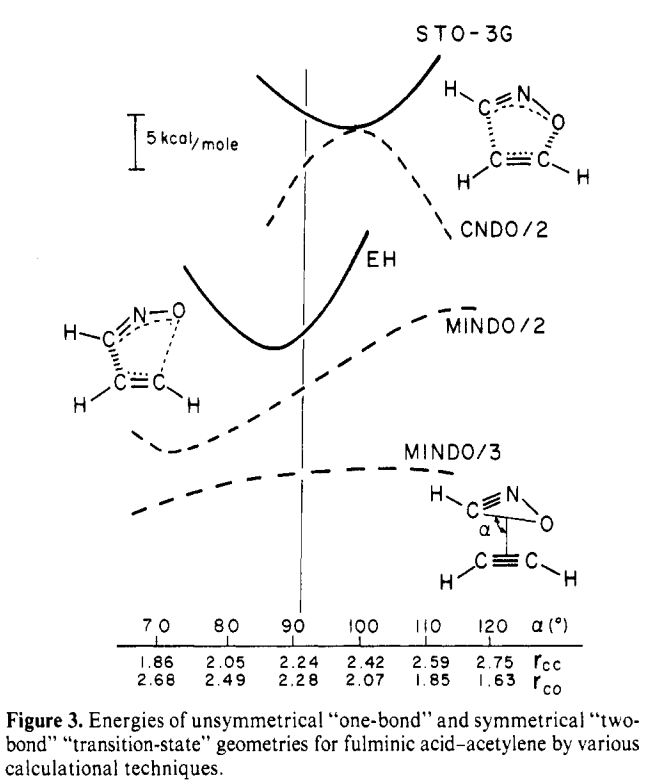
Moore’s Law is powerful. In the 1970s, using semiempirical methods and minimal basis set Hartree–Fock to investigate an 8-atom system was cutting-edge, as demonstrated by this paper from Ken Houk:
Figure 3 from the 1977 Houk paper.
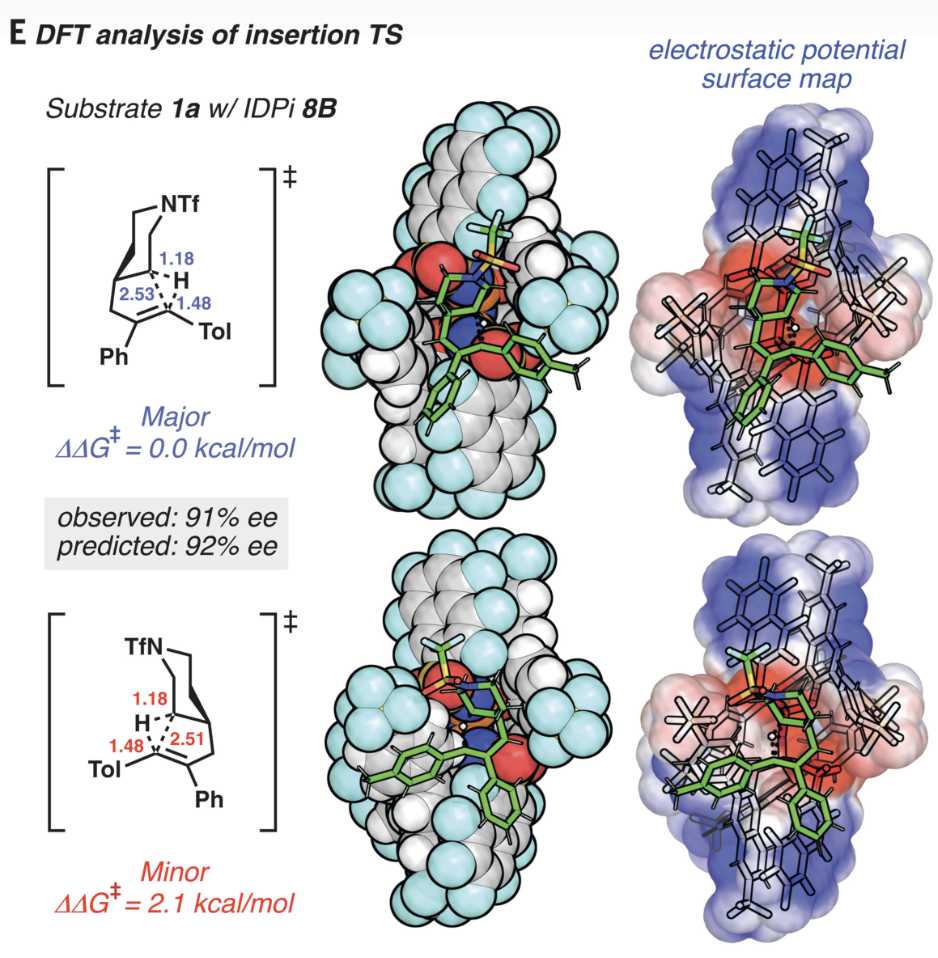
Now, that calculation would probably take only a few seconds on my laptop computer, and it’s becoming increasingly routine to perform full density-functional theory or post-Hartree–Fock studies on 200+ atom systems. A recent paper, also from Ken Houk, illustrates this nicely:
Figure 3E from the 2022 Houk paper.
The fact that it’s now routine to perform conformational searches and high-accuracy quantum chemical calculations on catalyst•substrate complexes with this degree of flexibility would astonish anyone from the past. (To be fair to computational chemists, it’s not all Moore’s Law—advances in QM tooling also play a big role.)
What Now?
There are lots of advances that I haven’t even covered, like the general improvement in synthetic methodology and the rise of robotics. Nevertheless, I think the trend is clear: it’s easier to acquire data than it’s ever been.
What, then, do we do with all this data? Most of the time, the answer seems to be “not much.” A recent editorial by Marisa Kozlowski observes that the average number of substrates in Organic Letters has increased from 17 in 2009 to 40 in 2021, even as the information contained in these papers has largely remained constant. Filling a substrate scope with almost identical compounds is a boring way to use more data; we can do better.
The availability of cheap data means that scientists can—and must—start thinking about new ways to approach experimental design. Lots of academic scientists still labor under the delusion that “hypothesis-driven science” is somehow superior to HTE, when in fact the two are ideal complements to one another. “Thinking in 96-well plates” is already common in industry, and should become more common in academia; why run a single reaction when you can run a whole screen?
New tools are needed to design panels, run reactions, and analyze the resultant data. One nice entry into this space is Tim Cernak’s phactor, a software package for high-throughput experimentation, and I’m sure lots more tools will spring up in the years to come. (I’m also optimistic that multi-substrate screening, which we and others have used in “screening for generality,” will become more widely adopted as HTE becomes more commonplace.)
The real worry, though, is that we will refuse to change our paradigms and just use all these improvements to publish old-style papers faster. All the technological breakthroughs in the world won’t do anything to accelerate scientific progress if we refuse to open our eyes to the possibilities they create. If present trends continue, it may be possible in 5–10 years to screen for a hit one week, optimize reaction conditions the next week, and run the substrate scope on the third week. Do we really want a world in which every methodology graduate student is expected to produce 10–12 low-quality papers per year?