Modeling ion-pair association/dissociation is an incredibly complex problem, and one that's often beyond the scope of conventional DFT-based techniques. (This study is a nice demonstration of how hard modeling ion-pairing can be.) Nevertheless, it's still often important to gain insight into what the relative energy of solvent-separated ion pairs and contact-ion pairs might be; are solvent-separated configurations energetically accessible or not?
I've run into this problem a few times myself: in measuring pKas in nonpolar solvents, and again recently when trying to understand the solution structure of ethereal HCl. When digging through the pKa literature, I was surprised to learn that there's a simple and relatively accurate way to estimate the dissociation constant of contact-ion pairs, developed by Raymond Fuoss in the 1950s.
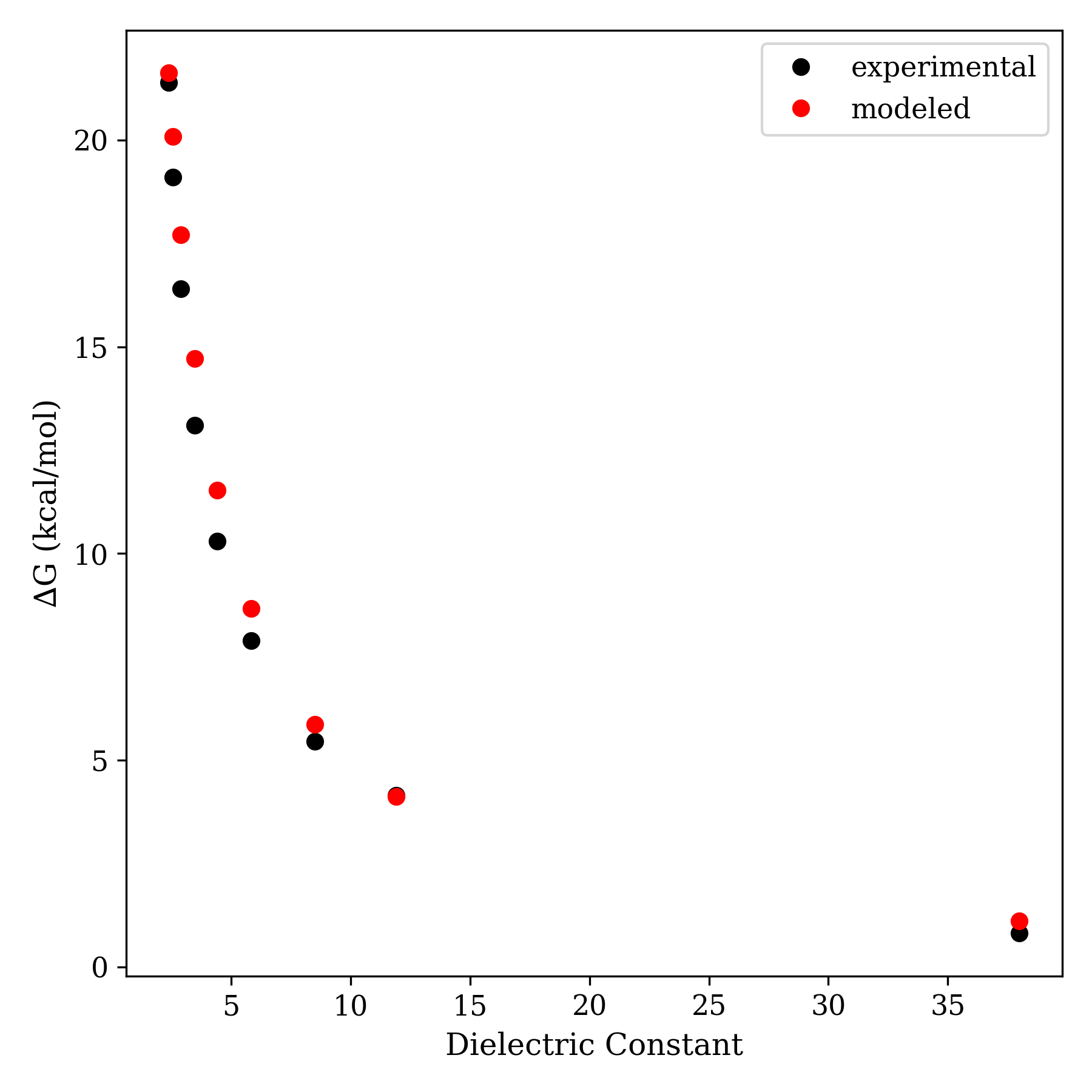
Despite modeling ions as charged spheres and solvent as a featureless dielectric, the "Fuoss model" is surprisingly good at reproducing experimental data. Here's one such example:
It's not trivial to think about how to get similar results using more atomistic methods! (In principle one could actually model the exact solvent mixture and compute the energy of ion-pair dissociation using biased sampling and MD, but this would be horrendously expensive and probably less accurate anyway.)
The Fuoss model is pretty simple to implement oneself—but to make things even easier, I've implemented it as an open-source Python package, which can be imported using pip. The package contains only a single function, compute_kd, which accepts the name of the cation, the name of the anion, and the dielectric constant of the medium. (Alternatively, .xyz files, .gjf files, or cctk.Molecule objects can also be given.)
Under the hood, the program builds molecules using cctk, computes their volume, and then applies the Fuoss model. The end result is a comically simple interface:
$ pip install quick_fuoss
$ python
>>> import quick_fuoss
>>> quick_fuoss.compute_kd("sodium", "chloride", 80)
1.0793241279015366
On the associated Github repository, there's also a little command-line script which makes this even simpler:
$ python quick_fuoss.py tetraisoamylammonium nitrate 8.5 Reading ion #1 from rdkit... Reading ion #2 from rdkit... Dissociation constant: 0.00004930 M Ionization energy: 5.873 kcal/mol $ python quick_fuoss.py tetraisoamylammonium nitrate 11.9 Reading ion #1 from rdkit... Reading ion #2 from rdkit... Dissociation constant: 0.00094706 M Ionization energy: 4.122 kcal/mol
My hope is that this program promotes wider adoption of the Fuoss model, and in general enables more critical thinking about ion-pair energetics in organic solvents. Please feel free to send any bug reports, complaints, etc. my way!