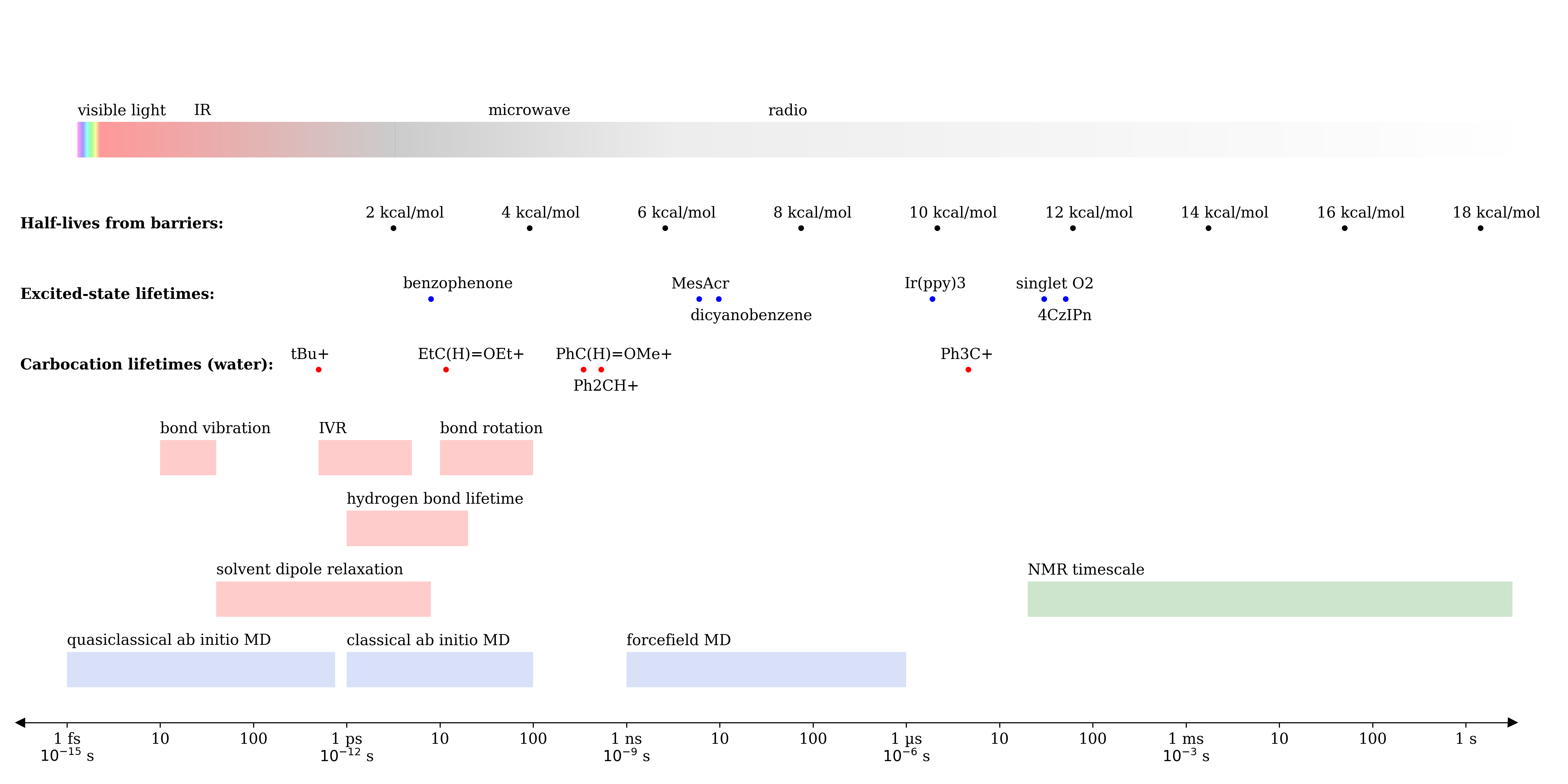
For many organic chemists, it’s hard to grasp the vast difference between various “fast” units of time. For instance, if a reactive intermediate has a lifetime of microseconds, does that mean it can escape the solvent shell and react with a substrate? What about a lifetime of nanoseconds, picoseconds, or femtoseconds?
To help answer these questions for myself, I made the following graphic about a year ago, which compares the timescale of various molecular processes on a logarithmic axis. Although someone skilled in Adobe Illustrator could doubtless make a prettier version, I've still found this to be a useful reference, and frequently use it as a slide in talks or group meetings:
Based on this graphic, it becomes easier to think about the interplay between competing fast processes. Species that persist for less than ~5 ps, for instance, are effectively “frozen” in the solvent configuration they’re formed in, whereas molecules that persist for longer can sample different solvent configurations. If a species can persist for 10-100 ps, it can begin to sample the nearby conformational landscape through low-barrier processes (e.g. single-bond rotation), although larger conformational changes might still be inaccessible.
As lifetimes stretch into the nanosecond regime, diffusion and solvent-cage escape become more realistic possibilities: based on Mayr’s value for the “diffusion limit” (2–4e9 M-1s-1), we can estimate that bimolecular association with a 1.0 M reactant will take 200-400 ps, while association with a 100 mM reactant will take 2-4 ns. On the far right of the graph, being able to distinguish different species by NMR (e.g. amide methyl groups in DMF) means that these species have a very long lifetime indeed.
Framed in these terms, then, it becomes obvious why the 10-100 ps timescales currently accessible by ab initio molecular dynamics (AIMD) are unable to model many important molecular processes. Indeed, work from Grossman and coworkers has shown that the results of AIMD simulations can be very dependent on the pre-AIMD equilibration method used, since the actual solvent environment is unable to fully relax over the timescale of the simulation. For AIMD to become a truly useful alternative to forcefield molecular dynamics, much faster ab initio methods will be needed!